
The Simultaneous Treatment of PC-3 Cells with the DNA-Demethylating Agent Decitabine and S-Adenosylmethionine Leads to Synergistic Anticancer Effects


Abstract
1. Introduction
2. Material and Methods
2.1. Cell Culture and Treatment
2.2. Proliferation Assay
2.3. Invasion and Migration Assay
2.4. Quantification of Global DNA Methylation
2.5. RNA Sequencing (RNA-seq)
2.6. Bioinformatic Analysis
2.7. Ingenuity Pathway Analysis (IPA) of Upstream Regulators
2.8. Promoter Methylation Analysis of Putative Proto-Oncogenes and Tumor-Suppressor Genes
3. Results
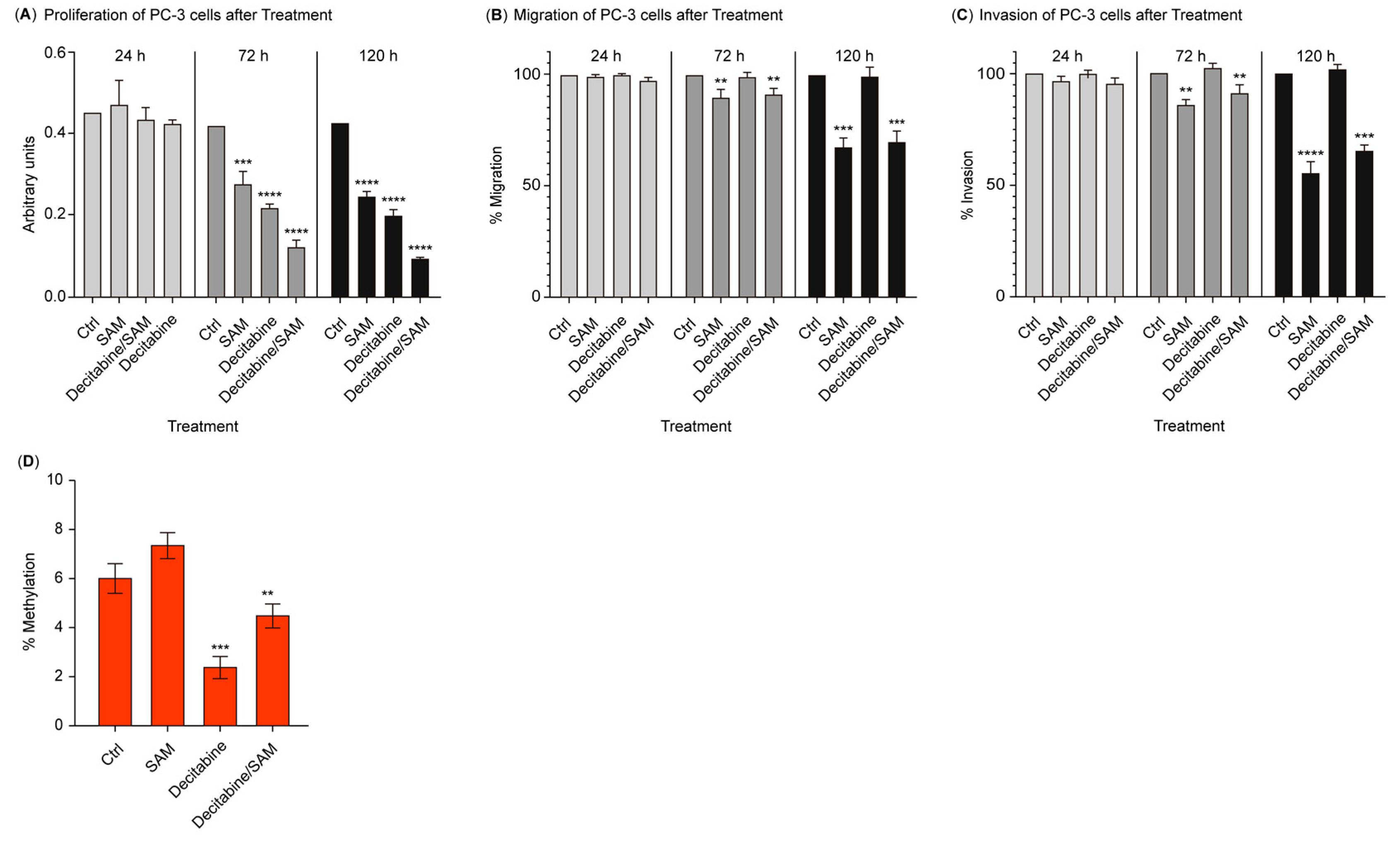
3.1. The Combination of SAM and Decitabine Synergizes the Suppression of Growth, Migration, and Invasion in PC-3 Cells
3.2. Treatment with SAM Prevents Global Hypomethylation Induced by Decitabine
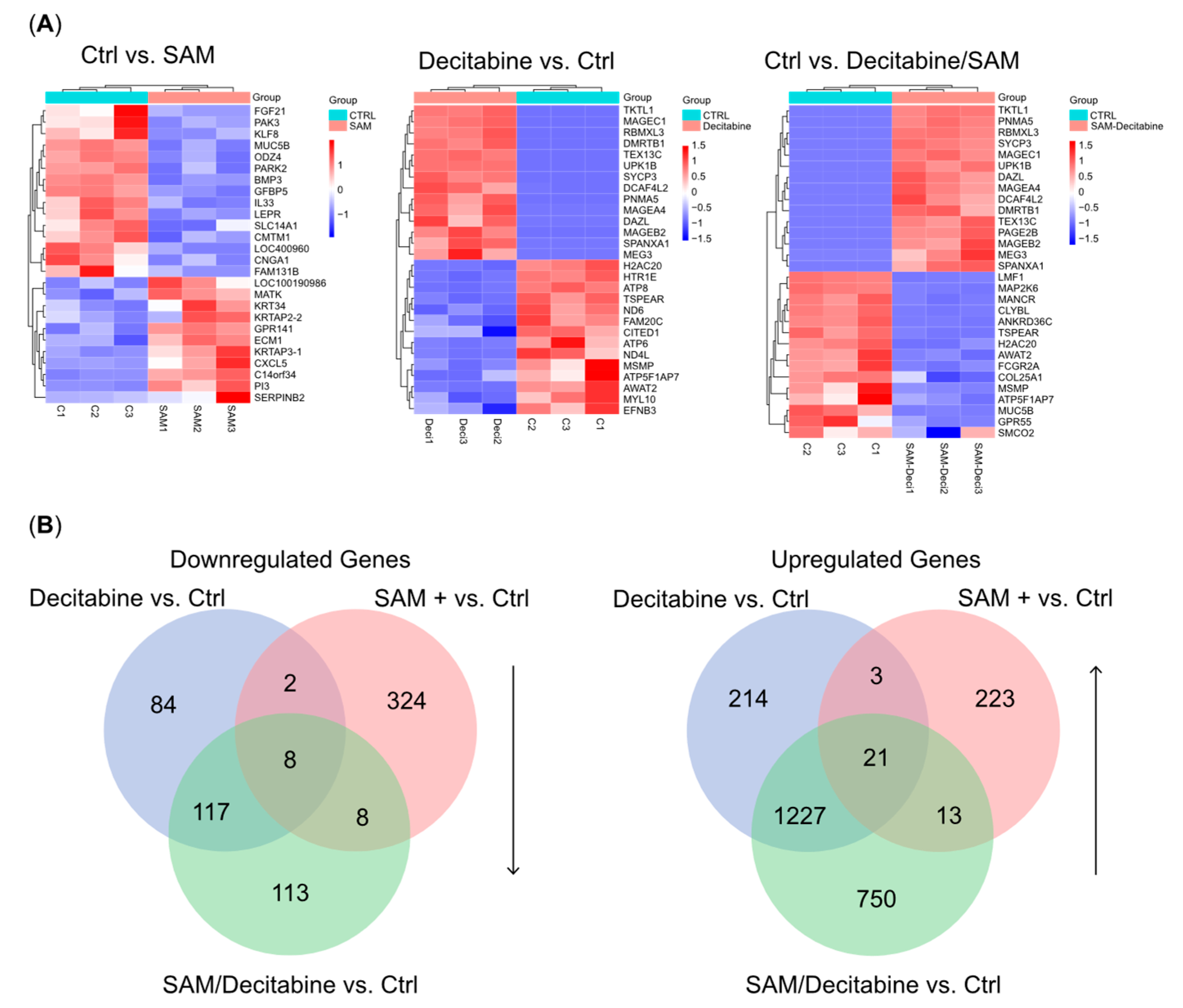
3.3. The Combination Treatment of SAM and Decitabine Affects the Transcriptome of PC-3 Cells
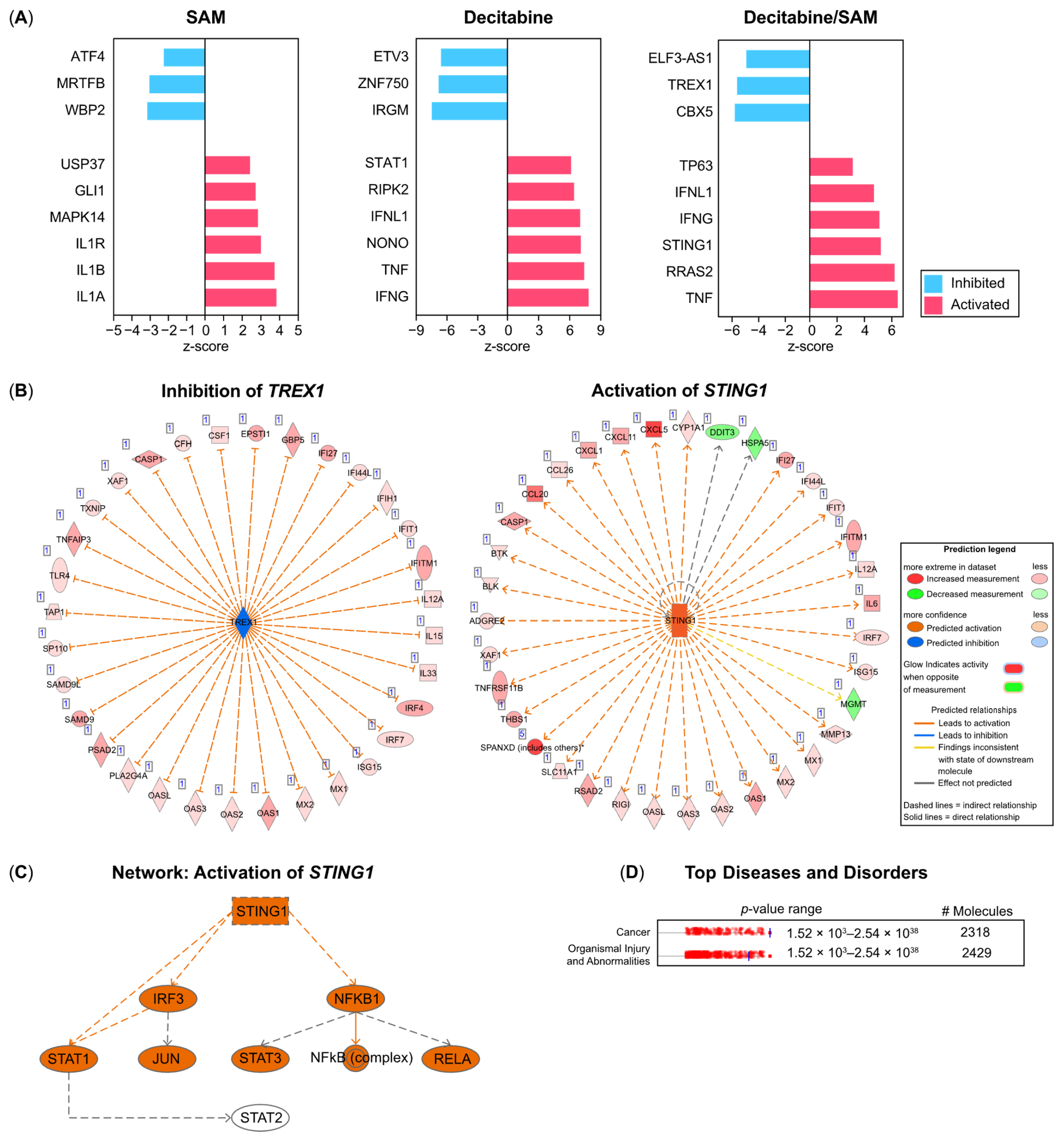
3.4. IPA Identifies Three Prime Repair Exonuclease (TREX)1, Stimulators of Interferon Genes (STING)1, and TP63 as Upstream Regulators in PC-3 Cells Treated with Combination Therapy (SAM/Decitabine)
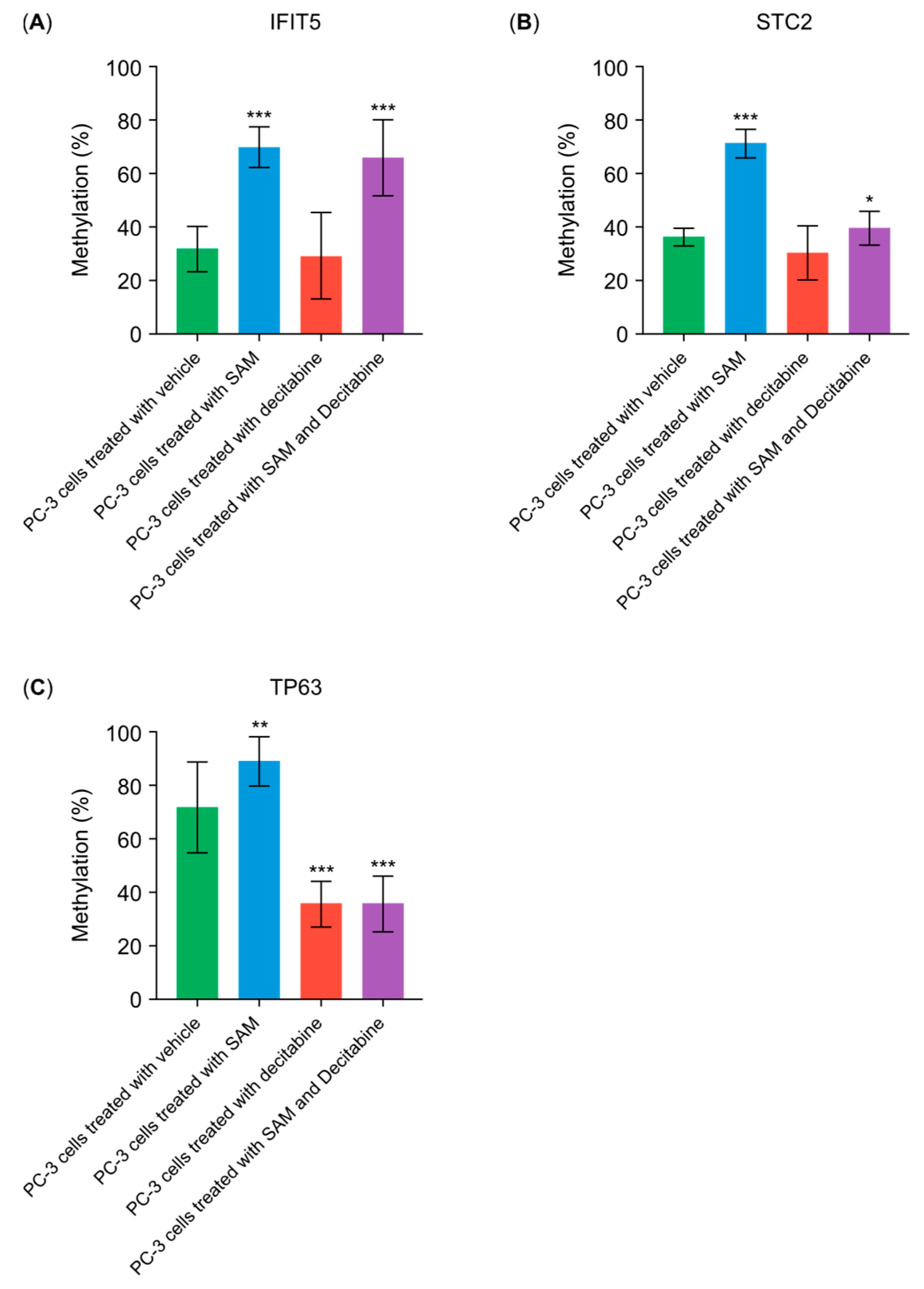
3.5. SAM Inhibits the Decitabine-Driven Hypomethylation in Proto-Oncogenes But Does Not Inhibit the Hypomethylation in Tumor-Suppressor Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ilango, S.; Paital, B.; Jayachandran, P.; Padma, P.R.; Nirmaladevi, R. Epigenetic alterations in cancer. Front. Biosci. 2020, 25, 1058–1109. [Google Scholar]
- Kanai, Y.; Hirohashi, S. Alterations of DNA methylation associated with abnormalities of DNA methyltransferases in human cancers during transition from a precancerous to a malignant state. Carcinogenesis 2007, 28, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.; An, J.; Ko, M. Epigenetic Regulators of DNA Cytosine Modification: Promising Targets for Cancer Therapy. Biomedicines 2023, 11, 654. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Loo, Y.H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Schmidt, T.; Leha, A.; Salinas-Riester, G. Treatment of prostate cancer cells with S-adenosylmethionine leads to genome-wide alterations in transcription profiles. Gene 2016, 595, 161–167. [Google Scholar] [CrossRef]
- Mathes, A.; Duman, M.B.; Neumann, A.; Dobreva, G.; Schmidt, T. S-adenosylmethionine treatment affects histone methylation in prostate cancer cells. Gene 2024, 893, 147915. [Google Scholar] [CrossRef]
- Shukeir, N.; Pakneshan, P.; Chen, G.; Szyf, M.; Rabbani, S.A. Alteration of the methylation status of tumor-promoting genes decreases prostate cancer cell invasiveness and tumorigenesis in vitro and in vivo. Cancer Res. 2006, 66, 9202–9210. [Google Scholar] [CrossRef]
- Fetrow, C.W.; Avila, J.R. Efficacy of the dietary supplement S-adenosyl-L-methionine. Ann. Pharmacother. 2001, 35, 1414–1425. [Google Scholar] [CrossRef]
- Stramentinoli, G.; Gualano, M.; Galli-Kienle, M. Intestinal absorption of S-adenosl-L-methionine. J. Pharmacol. Exp. Ther. 1979, 209, 323–326. [Google Scholar]
- Noureddin, M.; Sander-Struckmeier, S.; Mato, J.M. Early treatment efficacy of S-adenosylmethionine in patients with intrahepatic cholestasis: A systematic review. World J. Hepatol. 2020, 12, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Cahill, C.M.; Huang, X.; Roffman, J.L.; Lamon-Fava, S.; Fava, M.; Mischoulon, D.; Rogers, J.T. S-Adenosyl Methionine and Transmethylation Pathways in Neuropsychiatric Diseases Throughout Life. Neurotherapeutics 2018, 15, 156–175. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Higgins, D.P.; Yadav, D.K.; Godbole, A.A.; Pukkila-Worley, R.; Walker, A.K. Stress-responsive and metabolic gene regulation is altered in low S-adenosylmethionine. PLoS Genet. 2018, 14, e1007812. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Arakelian, A.; Cheishvili, D.; Szyf, M.; Rabbani, S.A. S-adenosylmethionine in combination with decitabine shows enhanced anti-cancer effects in repressing breast cancer growth and metastasis. J. Cell Mol. Med. 2020, 24, 10322–10337. [Google Scholar] [CrossRef]
- Chik, F.; Machnes, Z.; Szyf, M. Synergistic anti-breast cancer effect of a combined treatment with the methyl donor S-adenosylmethionine and the DNA methylation inhibitor 5-aza-2′-deoxycytidine. Carcinogenesis 2014, 35, 138–144. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Sergushichev, A.A.; Loboda, A.A.; Jha, A.K.; Vincent, E.E.; Driggers, E.M.; Jones, R.G.; Pearce, E.J.; Artyomov, M.N. GAM: A web-service for integrated transcriptional and metabolic network analysis. Nucleic Acids Res. 2016, 44, W194–W200. [Google Scholar] [CrossRef]
- Geistlinger, L.; Csaba, G.; Zimmer, R. Bioconductor’s EnrichmentBrowser: Seamless navigation through combined results of set- & network-based enrichment analysis. BMC Bioinform. 2016, 17, 45. [Google Scholar]
- Ye, C.; Jiang, N.; Zheng, J.; Zhang, S.; Zhang, J.; Zhou, J. Epigenetic therapy: Research progress of decitabine in the treatment of solid tumors. Biochim. Biophys. Acta Rev. Cancer 2023, 1879, 189066. [Google Scholar] [CrossRef]
- Lin, S.R.; Wen, Y.C.; Yeh, H.L.; Jiang, K.C.; Chen, W.H.; Mokgautsi, N.; Huang, J.; Chen, W.Y.; Liu, Y.N. EGFR-upregulated LIFR promotes SUCLG2-dependent castration resistance and neuroendocrine differentiation of prostate cancer. Oncogene 2020, 39, 6757–6775. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.H.; Yu, Y.P.; Zheng, Z.L.; Song, Y.; Xiang, G.S.; Nelson, J.; Michalopoulos, G.; Luo, J.H. Integrin alpha 7 interacts with high temperature requirement A2 (HtrA2) to induce prostate cancer cell death. Am. J. Pathol. 2010, 177, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Rodriguez, R.; Williams, K.; Silva, J.; Gutierrez, A.G.; Tyler, P.; Baharom, F.; Sun, T.; Lin, E.; Martin, S.; et al. The Exonuclease TREX1 Constitutes an Innate Immune Checkpoint Limiting cGAS/STING-Mediated Antitumor Immunity. Cancer Immunol. Res. 2024, 12, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Técher, H.; Gopaul, D.; Heuzé, J.; Bouzalmad, N.; Leray, B.; Vernet, A.; Mettling, C.; Moreaux, J.; Pasero, P.; Lin, Y.L. MRE11 and TREX1 control senescence by coordinating replication stress and interferon signaling. Nat. Commun. 2024, 15, 5423. [Google Scholar] [CrossRef]
- Miller, K.N.; Victorelli, S.G.; Salmonowicz, H.; Dasgupta, N.; Liu, T.; Passos, J.F.; Adams, P.D. Cytoplasmic DNA: Sources, sensing, and role in aging and disease. Cell 2021, 184, 5506–5526. [Google Scholar] [CrossRef]
- Tani, T.; Mathsyaraja, H.; Campisi, M.; Li, Z.H.; Haratani, K.; Fahey, C.G.; Ota, K.; Mahadevan, N.R.; Shi, Y.; Saito, S.; et al. TREX1 Inactivation Unleashes Cancer Cell STING-Interferon Signaling and Promotes Antitumor Immunity. Cancer Discov. 2024, 14, 752–765. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Grisanzio, C.; Signoretti, S. p63 in prostate biology and pathology. J. Cell Biochem. 2008, 103, 1354–1368. [Google Scholar] [CrossRef]
- Tucci, P.; Agostini, M.; Grespi, F.; Markert, E.K.; Terrinoni, A.; Vousden, K.H.; Muller, P.A.; Dötsch, V.; Kehrloesser, S.; Sayan, B.S.; et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 15312–15317. [Google Scholar] [CrossRef]
- Lo, U.G.; Pong, R.C.; Yang, D.; Gandee, L.; Hernandez, E.; Dang, A.; Lin, C.J.; Santoyo, J.; Ma, S.; Sonavane, R.; et al. IFNγ-Induced IFIT5 Promotes Epithelial-to-Mesenchymal Transition in Prostate Cancer via miRNA Processing. Cancer Res. 2019, 79, 1098–1112. [Google Scholar] [CrossRef]
- Tamura, K.; Furihata, M.; Chung, S.Y.; Uemura, M.; Yoshioka, H.; Iiyama, T.; Ashida, S.; Nasu, Y.; Fujioka, T.; Shuin, T.; et al. Stanniocalcin 2 overexpression in castration-resistant prostate cancer and aggressive prostate cancer. Cancer Sci. 2009, 100, 914–919. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer for Methyl-qPCR | Sequence (5′–3′) |
|---|---|
| STC fw | CCGACTCAGGAGAGCTC (Tm = 56 °C) |
| STC rev | CCCAGCCGTGTCACATG (Tm = 56 °C) |
| IFIT5 fw | GAAGCAGGGACTTAAGTTTC (Tm = 58 °C) |
| IFIT5 rev | GCTCTTGAGCTCTTCTATTAA (Tm = 58 °C) |
| TP63 | ACAACAGTAGAGAGGATGCC (Tm = 57 °C) |
| TP63 | CTCAAACTTACACTGTATTGA (Tm = 56 °C) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, T.; Sticht, C. The Simultaneous Treatment of PC-3 Cells with the DNA-Demethylating Agent Decitabine and S-Adenosylmethionine Leads to Synergistic Anticancer Effects. Genes 2024, 15, 1634. https://doi.org/10.3390/genes15121634
Schmidt T, Sticht C. The Simultaneous Treatment of PC-3 Cells with the DNA-Demethylating Agent Decitabine and S-Adenosylmethionine Leads to Synergistic Anticancer Effects. Genes. 2024; 15(12):1634. https://doi.org/10.3390/genes15121634
Chicago/Turabian StyleSchmidt, Thomas, and Carsten Sticht. 2024. "The Simultaneous Treatment of PC-3 Cells with the DNA-Demethylating Agent Decitabine and S-Adenosylmethionine Leads to Synergistic Anticancer Effects" Genes 15, no. 12: 1634. https://doi.org/10.3390/genes15121634
APA StyleSchmidt, T., & Sticht, C. (2024). The Simultaneous Treatment of PC-3 Cells with the DNA-Demethylating Agent Decitabine and S-Adenosylmethionine Leads to Synergistic Anticancer Effects. Genes, 15(12), 1634. https://doi.org/10.3390/genes15121634