1. Introduction
Peanuts (
A. hypogaea L.) hold a prominent position among oilseed crops due to their numerous nutritional attributes. They are widely recognized as an excellent source of edible oil, providing a high-quality oil that is widely used in cooking and food preparation. In addition to their oil content, peanuts are also valued for their protein content. Furthermore, peanuts offer dietary fiber, which aids in proper digestion and therefore is considered crucial for digestive health. Peanuts pack a range of essential minerals and vitamins, including magnesium, phosphorus, potassium, and B vitamins like niacin and folate. These nutrients play vital roles in various bodily functions, such as energy production, bone health, and nerve function [
1]. Their shells are used in the animal feed, fuel, and fertilizer industries [
2]. The haulm, typically utilized as animal feed, serves the dual purpose of providing fodder while also contributing to nitrogen fixation in the soil. The nitrogen fixation capability of the haulm can range from 100 kg ha
−1 to 152 kg ha
−1 [
3]. Peanuts are grown in more than 100 countries worldwide, producing 53.9 million metric tons (mt) from 32.7 million ha area [
4]. The crop is grown commercially between 40° N and 40° S latitudes; the largest producer in the world is China (17.9 mt), followed by India (9.9 mt) and Nigeria (4.5 mt). Peanut production is confronting severe biotic and abiotic stresses due to climate change, which emphasizes the necessity of climate-resilient crop production considering global food security. Agronomic traits such as biomass, pod weight, seed weight, and shelling percentage largely influence the yield and play a major role in the domestication, breeding, and selection of new peanut cultivars [
5]. Cultivated peanut around the world has a narrow genetic base. The linkage drag of desirable and undesirable traits often imposes biological constraints in developing improved cultivars by conventional crossing and selection [
6]. The crucial factors that affect the yield are the 100-pod weight (HPW), 100-seed weight (HSW), haulm yield, and shelling percentage (SP) [
7]. Traditional breeding methods face challenges when dealing with these quantitative traits. Moreover, these traits are typically governed by several genes, each with a relatively modest impact, which adds to their complexity and makes the breeding process laborious and time-consuming. Utilizing genomics-assisted breeding (GAB) plays a vital role in enhancing peanut yields substantially. Through the application of GAB, researchers can pinpoint and choose particular genes linked to yield-attributing traits, facilitating the more efficient development of high-yielding varieties compared to relying solely on traditional breeding methods [
8].
Furthermore, genotype by environment interaction (G × E) plays a significant role in determining the cultivar performance, imposing constraints in identifying the traits needed to improve productivity. The last decade has witnessed an increased demand for peanuts compared to other oilseed crops due to the increased use of peanuts in confectionary, health-sensitive consumers, and benefits to traders. Notably, the phenotypic selection of lines having significantly higher seed weight is difficult in standing crops. The availability of linked markers to seed weight will unfurl the scope of marker-based early-generation selection. The sequenced diploid ancestors have provided insights into understanding the genome of cultivated type [
9]. Evolutionary studies report low levels of genetic variation and polymorphism between the two sub-genomes [
10,
11].
Over the past five years, the increasing availability of genomic resources for wild species in peanut has opened up new avenues for the exploitation of genetic potential [
12]. Several studies were carried out to construct a genetic map to identify quantitative trait loci (QTL) associated with yield and its attributing traits [
13,
14,
15]. To date, a few SSR-based genetic maps are available for peanuts [
16,
17,
18]. However, the use of SSR markers is time-consuming and labor-intensive. It has low throughput [
19], while the presence of abundant genome-wide single-nucleotide polymorphisms (SNPs) can be exploited for map construction and for identifying genomic regions that control target traits [
20]. Various approaches, including genome-wide association studies, GWASs [
21], bulked segregant analyses, BSAs [
16], and specific-locus amplified fragment sequencing, SLAF-seq [
22] have been experimented upon for the identification and narrowing of the genomic regions/QTLs associated with yield- and quality-related traits in peanuts [
5,
23]. Recently, it has been shown that the QTL-seq approach could help in identifying a 1.89 Mb region on chromosome B06 linked to seed weight [
24] and overlapped regions on A09 and B02 for shelling percentage [
25]. Similarly, the identification of 36 marker–trait associations (MTAs) for pod length, pod length–width ratio, and 100-pod weight [
21] and six QTLs for seed weight [
23] added significantly to the understanding of the genetic basis of these traits. A total of three overlapping QTL hotspots were identified for haulm weight, pod weight, 100-seed weight, and SP, indicating the significant impact of these traits on peanut yield [
26].
Additionally, mapping minor alleles and their interactions is key to understanding their role in genomic-assisted breeding for improving yield-related traits [
27]. Most studies have targeted only the additive effects of genetic components, whereas minor alleles and epistatic interactions have remained unaddressed. Apart from additive QTLs, the phenotype of a plant is also regulated by epistatic QTLs and, therefore, should be considered in QTL analysis studies [
28,
29]. The intricate polygenic nature of the yield-attributing traits, its low heritability, minor allele interactions, and the substantial G × E interactions pose limitations on developing high-yielding cultivars that can perform well across diverse locations [
30]. Therefore, the objective of the current study was to use a recombinant inbred line (RIL) population (Valencia-C × JUG-03) to address the minor alleles’ interactions and identify the genomic regions and candidate genes associated with yield- and quality-related traits. GBS-based genotyping data were used to construct a dense genetic map. The genetic map, genotyping data, and multi-environment phenotyping data were used to identify the genomic regions associated with yield- and quality-related traits in peanuts.
4. Discussion
On a global scale, the adverse effects of climate change on crop productivity are evident, as they amplify various biotic and abiotic stresses, highlighting the urgency to improve existing cultivars. Consequently, strategies to exploit genetic variation become essential for peanut breeding as resistance against biotic and abiotic stresses directly impact peanut production. The utilization of genomics helps in targeting complex traits such as yield for improvement and utilizing novel alleles from wild species. Key traits like pod and seed weight are directly reflective of yield and have been widely studied in peanuts and other crops [
48,
49,
50,
51]. Initially, five SSR markers associated with pod- and kernel-related traits were identified through bulk segregant analysis [
16]. Subsequently, in the F
2 population (Zhonghua 10 × ICG12625), twenty-four QTLs (PVE- 1.69–18.70%) for HPW, HSW, SP, main stem height, pod length, seed length, pod length, and pod width were identified [
52]. Additionally, for shelling percentage, 25 QTLs were identified in the RIL population (Yuanza 9102 × Xuzhou 68-4) [
53]. These findings not only offer insight into gene discoveries but also help in the identification of functional markers for breeding.
In this study, a wide range of yield- and quality-related traits was observed for the RILs evaluated in two different environments, confirming the existence of genetic variability for different yield- and quality-related traits in peanuts [
27,
52,
53]. The population exhibited significant variation among the lines for haulm yield, oil content, protein content, linoleic acid, and oleic acid, indicating high variability among the tested RILs for the respective traits. A G × E interaction study is typically conducted to assess the adaptability and stability of lines or cultivars across different environments for quantitative traits. The higher the environmental variance, the higher the differential expression of lines across the environments. The environments used in the current study showed the inconsistency of RILs for some traits. Information on G × E interaction for yield- and quality-related traits is essential to develop effective selection strategies to improve yield in variable environments. The significant G × E interaction for pod yield, HPW, HSW, SP, and SCMR in the present study indicates inconsistent lines across environments, which was reported earlier [
54], emphasizing the importance of examining the lines in different environments. The performance of RILs varied significantly between environments due to large G × E interactions for pod yield, HPW, HSW, SP, and SCMR.
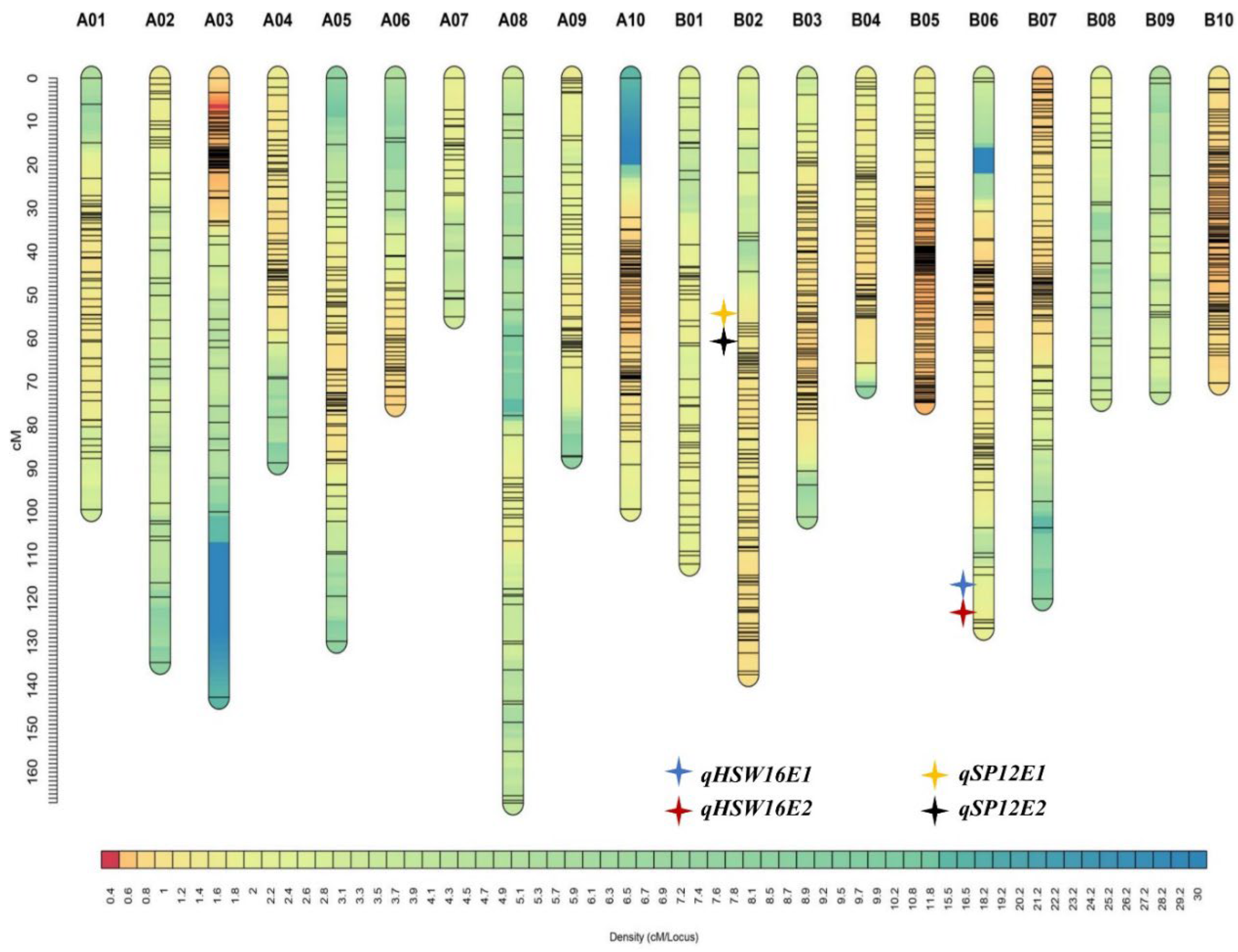
Yield-attributing traits have a complex interaction pattern, elucidated through the information of quantitative trait loci (QTL). These QTLs influence traits through cumulative effects. Moreover, epistatic interactions among minor loci affect multiple traits and must be incorporated along with QTLs’ introgression in the breeding program. Efforts were made to identify QTLs/genes associated with yield- and quality-related traits, followed by developing the lines by introgressing the selected genomic regions such as biotic and abiotic stresses through MAS [
55,
56,
57]. Using a high-density 58K “Axiom_
Arachis” array [
58] and RILs from a cross between TAG 24 and ICGV 86031, 1205 SNP loci spanning 2598.3 cM were mapped, with an average marker distance of 2.2 cM [
38]. The current work developed a linkage map comprising 1323 SNP loci, covering a total map length of 2003.13 cM, with an average marker distance of 1.89 cM between adjacent loci using the RIL mapping population. QTL analysis revealed that, except for four QTLs (two for HSW and two for SP), the phenotypic variance explained by the remaining QTL was <10%. It showed the complex nature of inheritance in peanuts. These observations support the previous report of multiple QTLs with minor effects associated with flowering date and maturity period in peanuts [
27]. Similarly, an RIL population (JH5 × M130) was used to construct a genetic map using 3130 markers, detecting QTLs for 100-pod weight and 100-seed weight on chromosomes A03, A04, A08, B04, B05, B06, and B08 of peanuts. A new genomic region of 0.36 Mb on chromosome A08 was detected as a hotspot, including 18 candidate genes [
48]. Moreover, the genomic regions for 100-seed weight and shelling percentage were also identified using the RIL population (Chico × ICGV 02251). QTL analysis identified three consistent QTLs on chromosomes A05, A08, and B10, whereas seven QTLs were found on chromosomes A01, A02, A04, A10, B05, B06, and B09 for 100-seed weight [
59]. In a study utilizing an RIL population derived from the cross between JH6 and KX01-6, two stable QTLs (
qHYF_A08 and
qHYF_B06) were identified across six different environments. The QTL
qHYF_A08 showed a predominant association with variations in shelling percentage and 100-pod weight, exhibiting PVE values ranging from 5.78% to 23.20%. Conversely,
qHYF_B06 was primarily linked to variations in 100-pod weight and 100-seed weight, with PVE values ranging from 13.38% to 31.29% [
60].
The phenomenon of consistent QTLs detected under different environments with significant G × E interaction was reported in peanuts [
61,
62]. Two major QTLs mapped for HSW and two for SP were on B06 and B02, respectively. Thus, chromosomes B02 and B06 harbored important regions for SP and HSW, respectively. Similarly, the major and consistent QTL,
cqSPB02, was identified on chromosome B02 for shelling percentage, with phenotypic variance explained being 10.47–17.01% across four different environments in the RIL population (Yuanza 9102 × Xuzhou 68-4) [
53]. Moreover, three major QTLs (
q100SW16a,
q100SW16a, and
q100SW16a) were found on chromosome B06 for 100-seed weight having PVE of 29.81–35.39% across four different seasons [
63], indicating stable genetic effects independent of environments. The primary influence of a QTL cannot be solely attributed to the genetic background; in certain cases, it could be influenced by environmental factors or a combination of both. One notable QTL for HSW was located on chromosome B05, as well as two significant QTLs for shelling percentage which were found on chromosomes B06 and B10, which demonstrated substantial additive effects influenced by the environment [
59].
The common QTL regions governing different traits suggest the relationship between these traits, pleiotropy effects, and/or tightly linked genes [
5]. The genomic region associated with HSW between the marker interval of S16_2332048 and S16_8231918 on chromosome B06 harbored ten genes. These genes encoded the protein kinase superfamily protein, transcription factor
bHLH68, the
GTP binding elongation factor Tu family protein, the
CBS domain-containing protein, the
ribosomal protein L19e family protein, the
seed maturation protein, the
ethylene-responsive transcription factor,
NAD + ADP-ribosyltransferase,
isopentenyltransferase and the
cytochrome P450 superfamily protein. Similarly, the genomic region associated with SP (S12_42838843-S12_73270208) on chromosome B02 harbored four genes. These genes encoded protein
MIZU-KUSSEI 1, the
actin-related protein,
serine/threonine-protein phosphatase and the
disease resistance protein (
TIR-NBS-LRR). Nine of the fourteen genes had annotations directly or indirectly related to yield-related traits.
The disease resistance protein coding gene (
Araip.6MG4Z) was highly expressed in seeds and detected in the major genomic region of shelling percentage (
Figure 4). This reveals that the disease resistance protein,
TIR-NBS-LRR, may be indirectly involved in pod and seed development in addition to providing resistance to the plant against disease infestation. This gene is a part of receptor-like kinase gene family, known for salt tolerance and low temperature resistance induced by ABA [
38]. The
serine-threonine phosphatase-encoding gene (
Araip.DH675) was highly expressed on pod walls and found to be associated with signal transduction for cell division and differentiation. The
serine-threonine phosphatase protein-encoding gene was also reported in the B02 chromosome associated with shelling percentage in peanuts [
61]. In rice, the
serine-threonine phosphatase gene contains the Kelch motif, which determines the larger grain size and thus contributes to yield increment [
64]. Similarly, the
serine-threonine phosphatase gene also identified in the major QTL region was associated with pod length in soybean [
65]. In maize, the role of the
serine/threonine protein kinase-encoding gene
KNR6 was reported for ear length, and the overexpression of this genomic region resulted in significantly increased yield [
66].
The gene protein kinase superfamily (
Araip.49T7Y) is expressed in cotyledons and seeds. A kinase protein such as mitogen-activated protein kinase (MPK3) regulates the mitotic activities in the integumental cells through phosphorylation. These proteins may be involved in pod and seed development through protein–protein interactions [
67]. The role of
calcium dependent protein kinase (
CDPK) was evaluated in developing peanut pods [
68]. The higher expression of CDPK in early pod development might suggest that the absorption of Ca
2+ occurs directly through the epidermal layer of pods in addition to via the xylem route. This argument was supported by the transcriptional upregulation of CDPK only in the development of seeds in Ca
2+-deficient zones [
68]. The
seed maturation protein coding gene (
Araip.GWR7V) is expressed higher in seed tissues than other vegetative tissues, indicating its role in seed maturation and development. The upregulation of the seed maturation protein [
69] in seed tissues is consistent with the high activity of protein synthesis in seeds. Genes related to seed maturation, such as those involved in the seed storage protein and the accumulation of lipids, are usually regulated by the interaction of cis-acting elements in the promoter region and transcriptional regulators [
70]. These regulatory networks promote the accumulation of seed storage reserves and thus lead to an increase in seed weight. Further, the expression pattern of the
Araip.GWR7V gene was preferentially higher in seeds, which indicated that the promoter region of
Araip.GWR7V can function in a seed-specific manner [
71]. The
isopentyltransferase (
IPT) gene (
Araip.UY42T) is one of the critical enzymes involved in cytokinin biosynthesis. The
IPT-expressing peanut plant was identified with higher biomass in a dryland condition in the field [
72]. This significant positive correlation of the higher yield of IPT-expressing plants with an increase in photosynthesis indicated the role of the cytokinin-mediated regulation of photosynthesis in transgenic plants. The differential expression pattern of the
IPT gene showed that the regulatory function of the IPT gene in cytokinesis biosynthesis was one of the prime factors for determining pod size in peanuts [
73]. The ethylene-responsive transcription factor (
Araip.LE5CL) is highly expressed in seeds, and previously, it was reported in associated genomic regions of haulm weight [
37]. The important role of ethylene-responsive transcription factors in the early development of peanut pods has also been identified [
74]. Generally, Ca
2+ ions promote pod maturation and development; however, the downregulation of genes encoding the ethylene-responsive transcription factor was reported in the presence of Ca
2+ [
75]. This indicated that ethylene-responsive transcription factors have a negative correlation with pod formation and development. Moreover, the ethylene-responsive element-binding factor family has a large number of transcription factors which are involved in abiotic and biotic stresses in plants [
76,
77]. The ethylene-responsive transcription factor superfamily genes, namely
GmAP2-1,
GmAP2-2,
GmAP2-3,
GmAP2-4,
GmAP2-5,
GmAP2-6, and
GmAP2-7, had important roles in the regulation of seed length and seed width in overexpressed transgenic lines of Arabidopsis [
78]. The members of the family of the
cytochrome P450 protein-encoding gene (
Araip.WM0UU) are involved in brassinosteroid biosynthesis. The
CYP72C1 gene (a
cytochrome P450 monooxygenase family) regulates cell elongation and therefore results in short petioles and shortened seeds along the longitudinal axis [
79]. This study supports that members of the
cytochrome P450 gene affect the seed size and its elongation by regulating the brassinosteroid level [
79]. The
CBS-domain-containing protein (
Araip.CXF88) was expressed at relatively higher levels in cotyledons than in other tissues (
Figure 4). Similarly, the expression of the CBS-domain-containing protein in cotyledons and floral tissues in addition to anthers was reported in the
proCBSX1:GUS-expressing transgenic line of
Arabidopsis [
80]. The overexpression of the
CBS-domain-containing protein was reported to increase the soybean’s low nitrogen stress tolerance [
81]. The
bHLH transcription factor (
Araip.5E3CZ) is specifically expressed only in cotyledon tissues, which may explain its role in seed development (
Figure 4). In addition, it also participates in other developmental processes such as the proper growth of axillary meristems, root hair, and anthers [
82]. The higher expression of
bHLH TFs in peanut seed tissues signifies its importance in seed development and maturation [
38,
83] and its pleiotropic role in plant growth and development as well as in stress responses [
84,
85]. Similarly, a bHLH transcription factor (TaPGS1) was specifically overexpressed in wheat and rice lines, which resulted in increased grain weight [
86]. In addition, a yeast one-hybrid assay showed that the overexpression of the bHLH transcription factor AhbHLH121 resulted in the increased activity of antioxidant enzymes under stress by facilitating the expression of the genes for peroxidase, catalase, and superoxide dismutase in peanuts [
87].
In addition, epistatic effects (the interaction of different loci in a population) play a significant role in determining trait expression [
88,
89]. A total of 91 pairs of QTL interactions were detected for all traits, suggesting that apart from environmental effects, epistatic QTLs also play a non-additive role in the inheritance of these traits. Such results are not surprising given that epistasis is more important for traits governed by several QTLs with small effects than for those governed by a few large major QTLs [
90]. Epistatic QTLs affecting more than one trait were also reported in peanuts for pod number per plant [
91]. Likewise, a total of 73 pairs of epistatic interactions involving 92 loci were discovered for pod length, pod width, length–width ratio, pod roundness, beak degree, and constriction degree. These interactions collectively accounted for phenotypic variations ranging from 0.94% to 6.45% [
92].


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}