
Genomic Features of Homologous Recombination Deficiency in Breast Cancer: Impact on Testing and Immunotherapy



Abstract
:Simple Summary
Abstract
1. Introduction
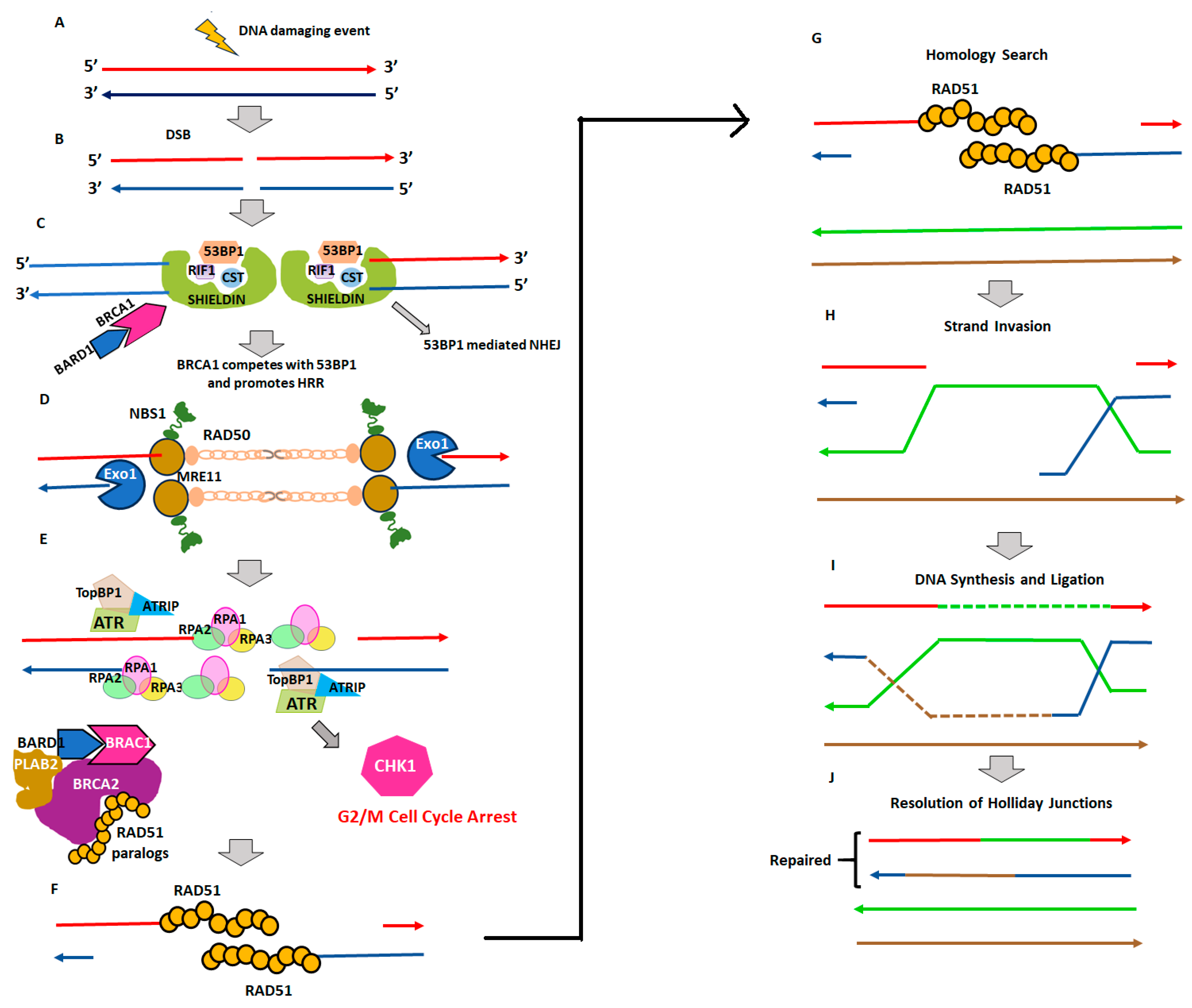
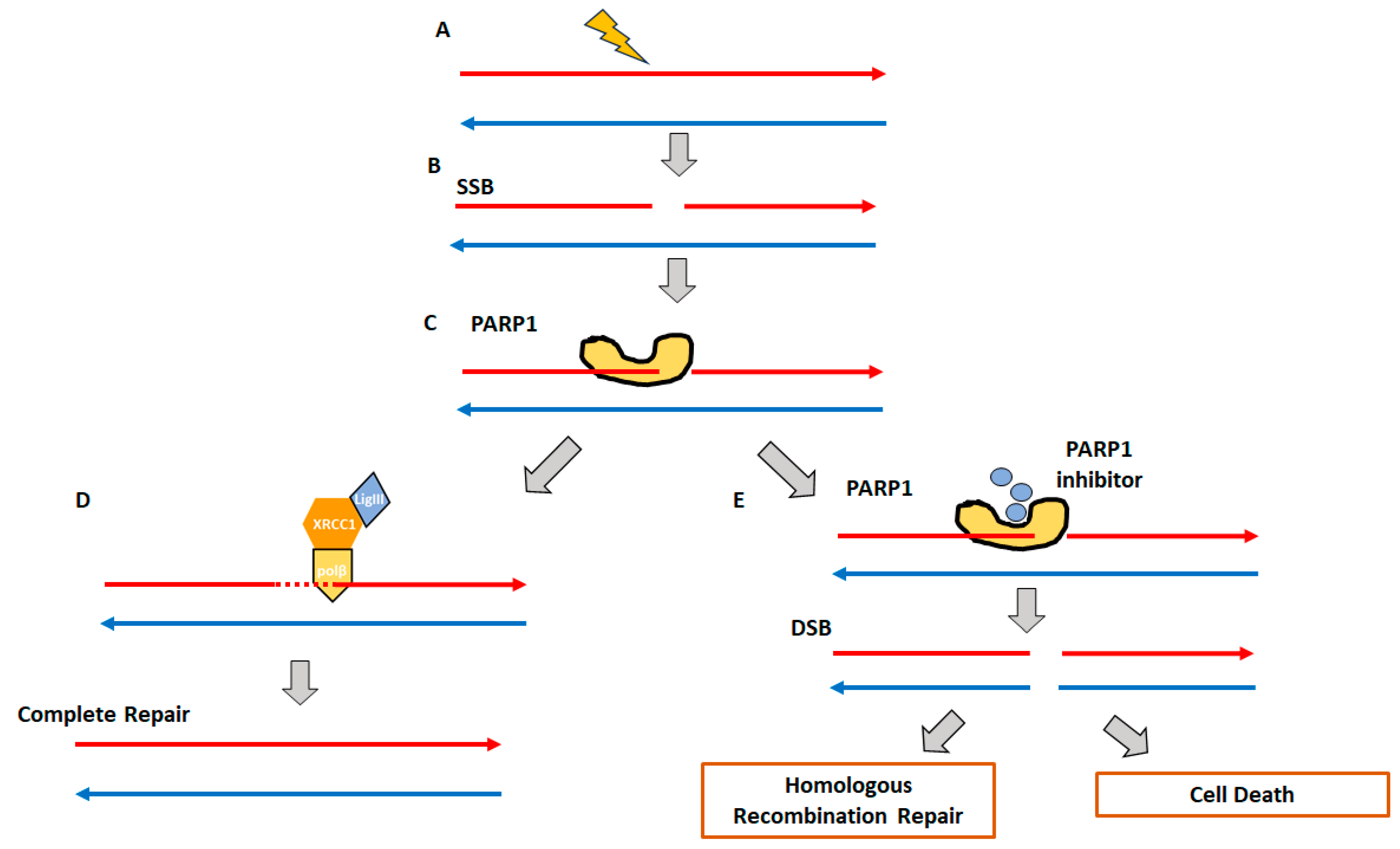
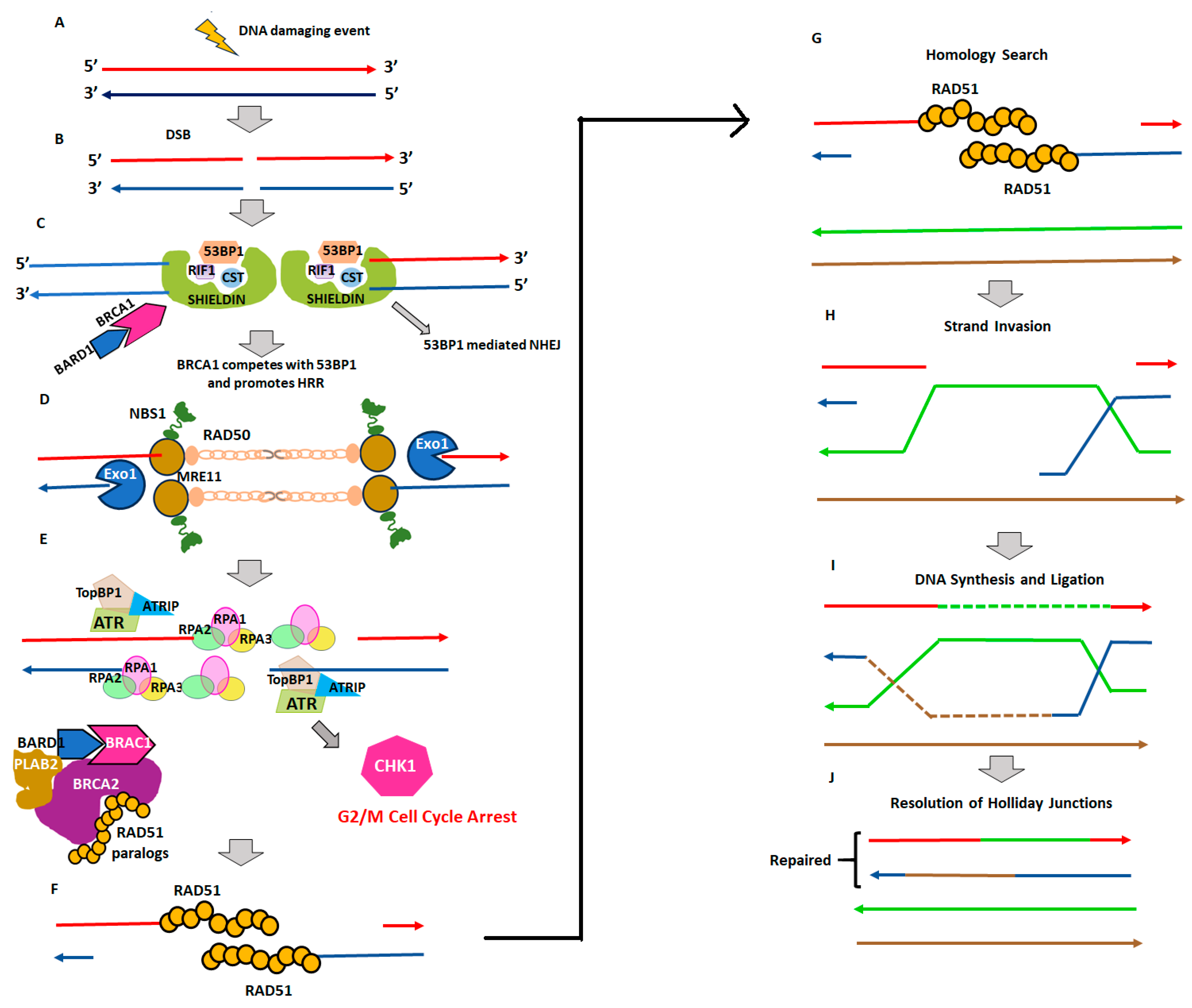
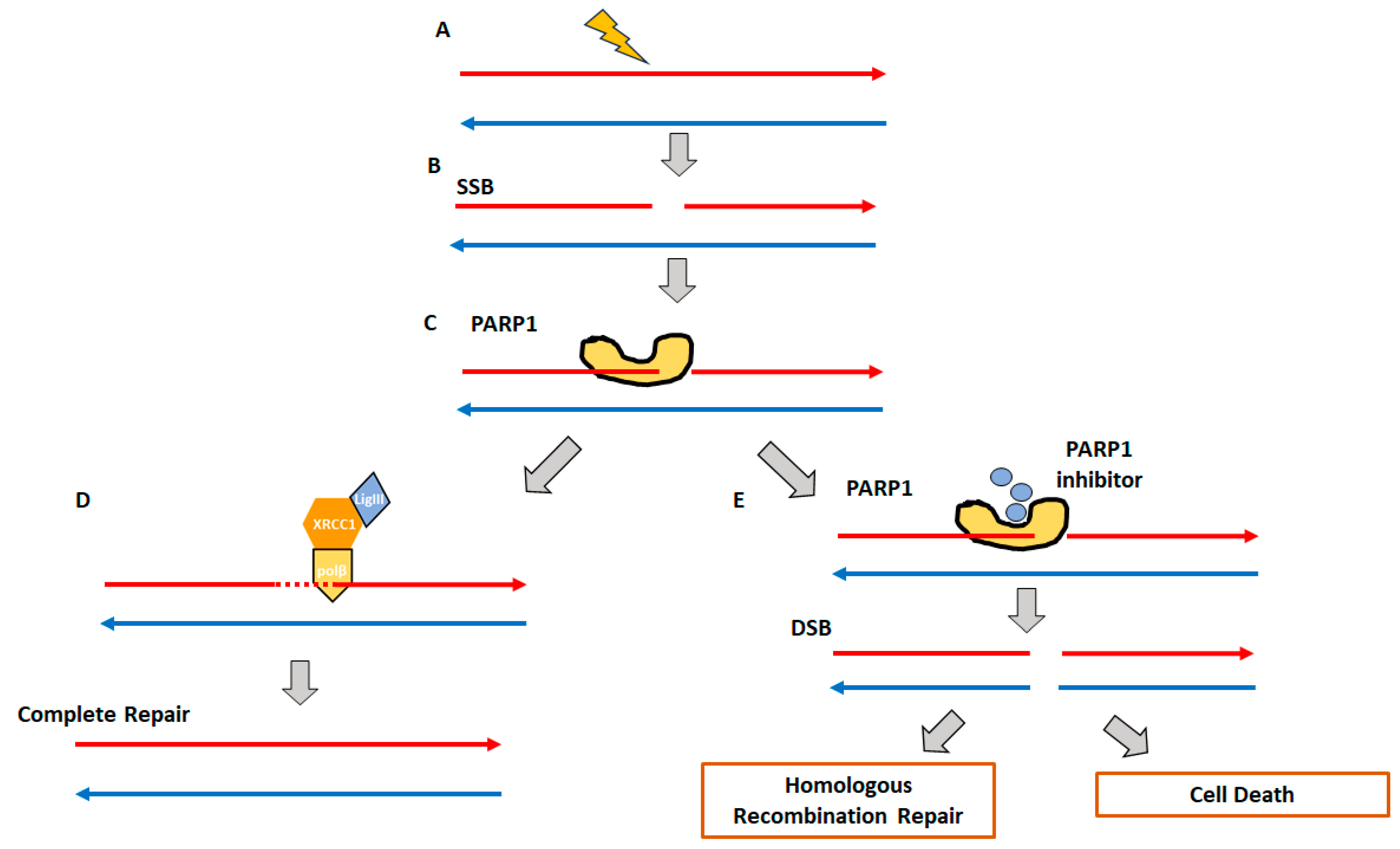
2. Homologous Recombination Deficiency and Genomic Scars
2.1. Homologous Recombination Deficiency (HRD) and BRCAness
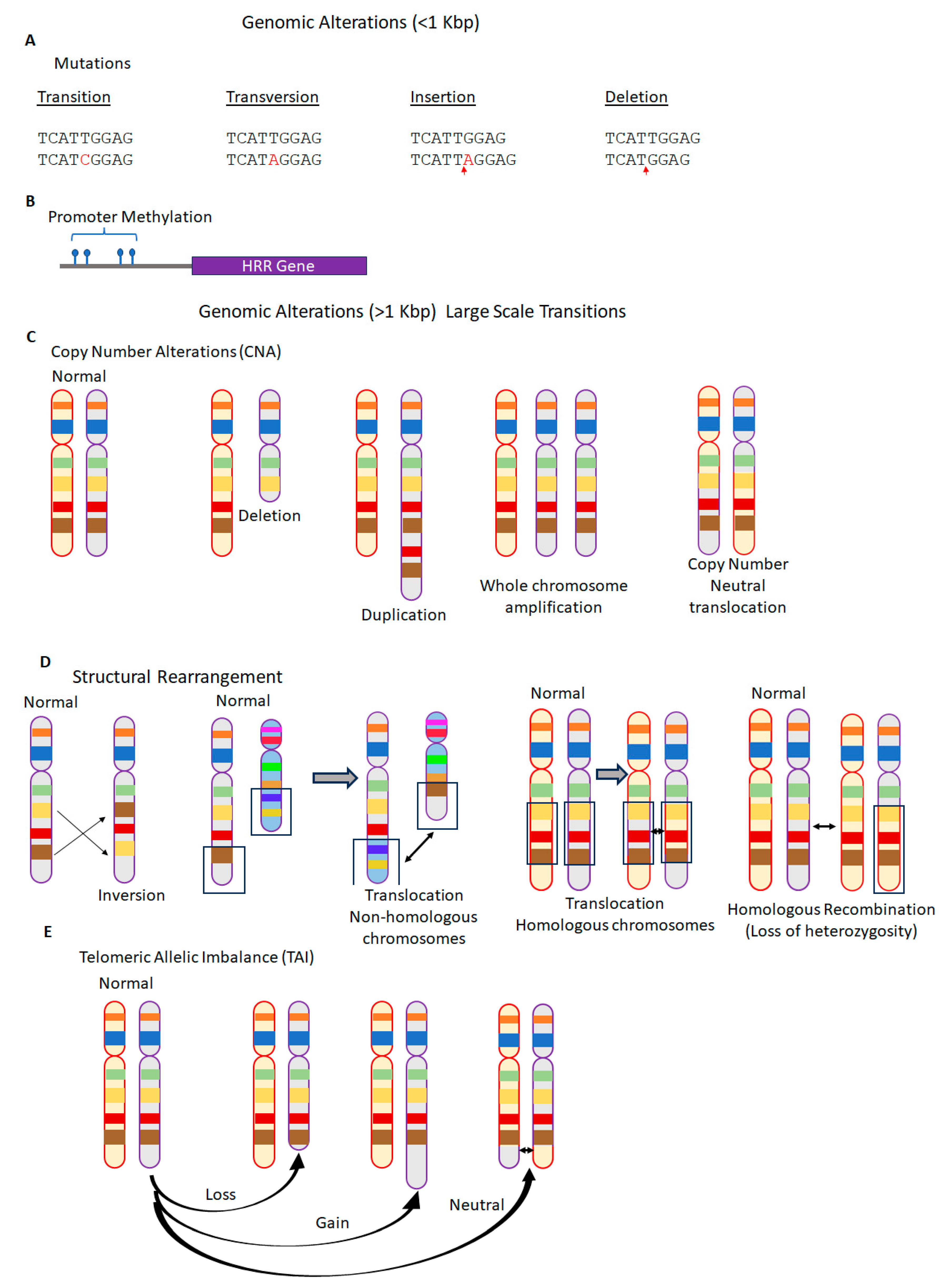
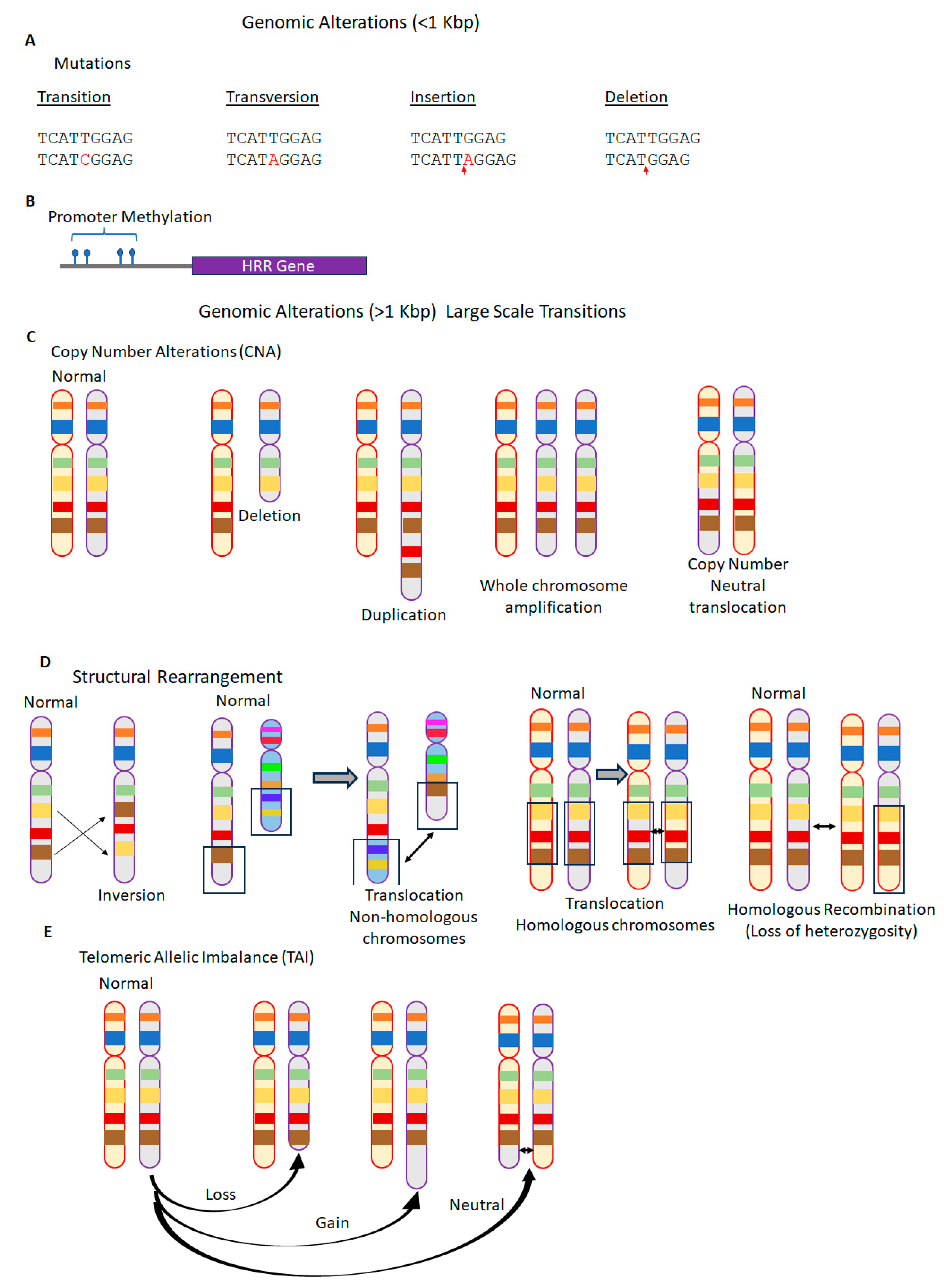
2.2. Genomic Instability and Genomic Scars
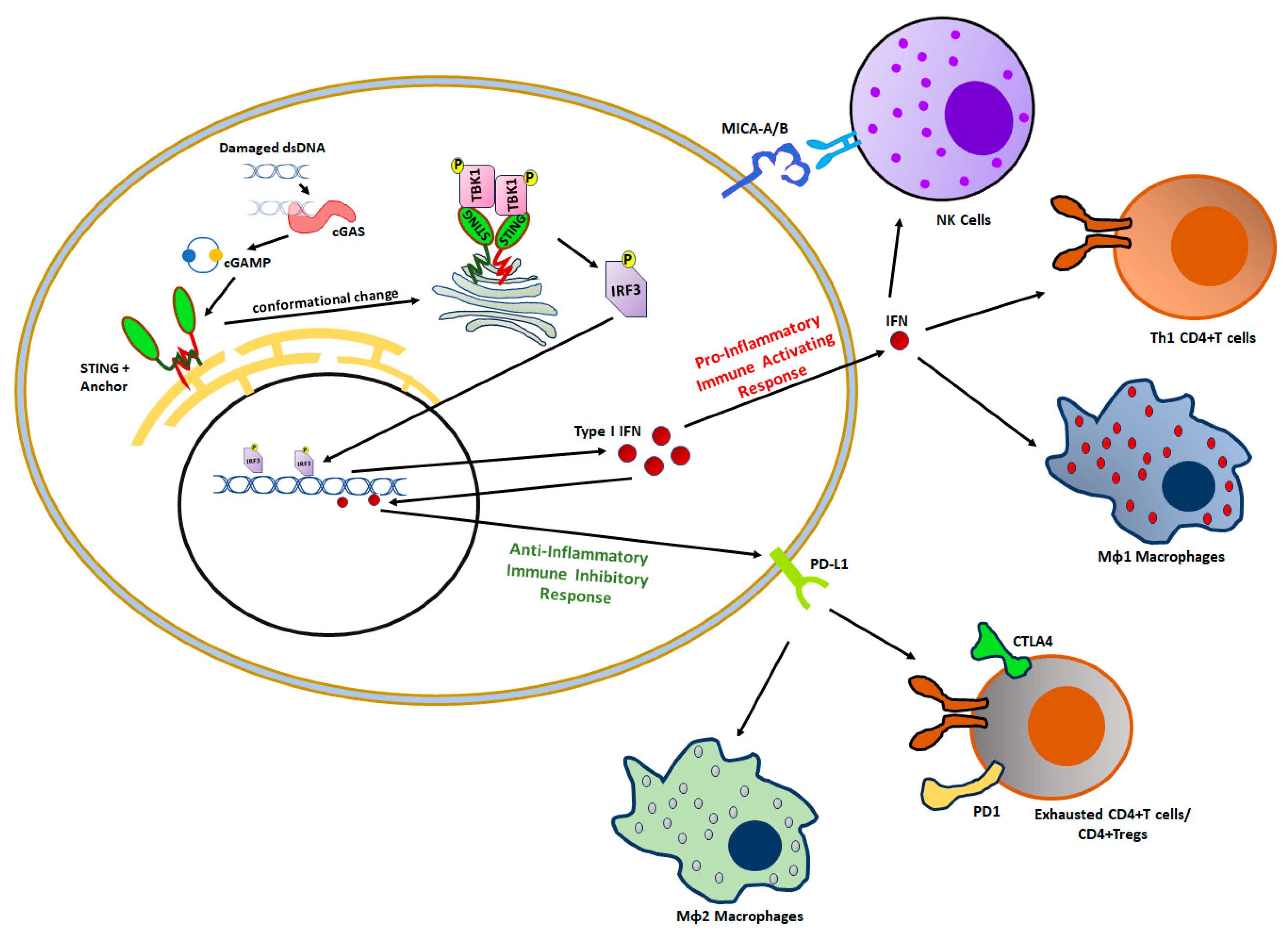
3. Impact of BRCAness on Tumor Immunity
3.1. Tumor-Infiltrating Innate Immune Cells
3.2. Tumor-Infiltrating Adaptive Immune Cells
4. Impact of BRCAness on Early Breast Cancer Treatment
5. Impact of BRCAness on Metastatic Breast Cancer Treatment
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Vitor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Chen, Y.; Zhang, D.; Wei, Y.; Li, Z.; Li, Q.; Xu, B. Homologous Recombination Deficiency (HRD) and BRCA 1/2 Gene Mutation for Predicting the Effect of Platinum-Based Neoadjuvant Chemotherapy of Early-Stage Triple-Negative Breast Cancer (TNBC): A Systematic Review and Meta-Analysis. J. Pers. Med. 2022, 12, 323. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef]
- Sharma, P.; Barlow, W.E.; Godwin, A.K.; Pathak, H.; Isakova, K.; Williams, D.; Timms, K.M.; Hartman, A.R.; Wenstrup, R.J.; Linden, H.M.; et al. Impact of homologous recombination deficiency biomarkers on outcomes in patients with triple-negative breast cancer treated with adjuvant doxorubicin and cyclophosphamide (SWOG S9313). Ann. Oncol. 2018, 29, 654. [Google Scholar] [CrossRef]
- Yndestad, S.; Engebrethsen, C.; Herencia-Ropero, A.; Nikolaienko, O.; Vintermyr, O.K.; Lillestol, R.K.; Minsaas, L.; Leirvaag, B.; Iversen, G.T.; Gilje, B.; et al. Homologous Recombination Deficiency across Subtypes of Primary Breast Cancer. JCO Precis. Oncol. 2023, 7, e2300338. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Galland, L.; Ballot, E.; Mananet, H.; Boidot, R.; Lecuelle, J.; Albuisson, J.; Arnould, L.; Desmoulins, I.; Mayeur, D.; Kaderbhai, C.; et al. Efficacy of platinum-based chemotherapy in metastatic breast cancer and HRD biomarkers: Utility of exome sequencing. NPJ Breast Cancer 2022, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage repair in cancer: From mechanisms to applications. Ann. Transl. Med. 2020, 8, 1685. [Google Scholar] [CrossRef]
- Liu, K.; Mao, X.; Li, T.; Xu, Z.; An, R. Immunotherapy and immunobiomarker in breast cancer: Current practice and future perspectives. Am. J. Cancer Res. 2022, 12, 3532. [Google Scholar]
- Das, S.; Johnson, D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef]
- Shi, W.J.; Zhao, W. Biomarkers or factors for predicting the efficacy and adverse effects of immune checkpoint inhibitors in lung cancer: Achievements and prospective. Chin. Med. J. 2020, 133, 2466. [Google Scholar] [CrossRef]
- Rizk, E.M.; Seffens, A.M.; Trager, M.H.; Moore, M.R.; Geskin, L.J.; Gartrell-Corrado, R.D.; Wong, W.; Saenger, Y.M. Biomarkers Predictive of Survival and Response to Immune Checkpoint Inhibitors in Melanoma. Am. J. Clin. Dermatol. 2020, 21, 1. [Google Scholar] [CrossRef]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2020, 8, 34. [Google Scholar] [CrossRef]
- Wang, P.; Chen, Y.; Wang, C. Beyond Tumor Mutation Burden: Tumor Neoantigen Burden as a Biomarker for Immunotherapy and Other Types of Therapy. Front. Oncol. 2021, 11, 672677. [Google Scholar] [CrossRef]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov. 2020, 10, 1808. [Google Scholar] [CrossRef]
- van Wilpe, S.; Tolmeijer, S.H.; Koornstra, R.H.T.; de Vries, I.J.M.; Gerritsen, W.R.; Ligtenberg, M.; Mehra, N. Homologous Recombination Repair Deficiency and Implications for Tumor Immunogenicity. Cancers 2021, 13, 2249. [Google Scholar] [CrossRef]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309. [Google Scholar] [CrossRef]
- Silva, S.B.; Wanderley, C.W.S.; Colli, L.M. Immune Checkpoint Inhibitors in Tumors Harboring Homologous Recombination Deficiency: Challenges in Attaining Efficacy. Front. Immunol. 2022, 13, 826577. [Google Scholar] [CrossRef]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Osterman, M.D.; Li, M. Tissue specificity of DNA damage response and tumorigenesis. Cancer Biol. Med. 2019, 16, 396. [Google Scholar] [CrossRef] [PubMed]
- Jachimowicz, R.D.; Goergens, J.; Reinhardt, H.C. DNA double-strand break repair pathway choice—From basic biology to clinical exploitation. Cell Cycle 2019, 18, 1423. [Google Scholar] [CrossRef]
- Elbakry, A.; Lobrich, M. Homologous Recombination Subpathways: A Tangle to Resolve. Front. Genet. 2021, 12, 723847. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef]
- Qi, L.; Chakravarthy, R.; Li, M.M.; Deng, C.X.; Li, R.; Hu, Y. Phosphorylation of BRCA1 by ATM upon double-strand breaks impacts ATM function in end-resection: A potential feedback loop. iScience 2022, 25, 104944. [Google Scholar] [CrossRef]
- Foo, T.K.; Vincelli, G.; Huselid, E.; Her, J.; Zheng, H.; Simhadri, S.; Wang, M.; Huo, Y.; Li, T.; Yu, X.; et al. ATR/ATM-Mediated Phosphorylation of BRCA1 T1394 Promotes Homologous Recombinational Repair and G(2)-M Checkpoint Maintenance. Cancer Res. 2021, 81, 4676. [Google Scholar] [CrossRef]
- Bonilla, B.; Hengel, S.R.; Grundy, M.K.; Bernstein, K.A. RAD51 Gene Family Structure and Function. Annu. Rev. Genet. 2020, 54, 25. [Google Scholar] [CrossRef]
- Holloman, W.K. Unraveling the mechanism of BRCA2 in homologous recombination. Nat. Struct. Mol. Biol. 2011, 18, 748. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524. [Google Scholar] [CrossRef]
- Song, Q.; Hu, Y.; Yin, A.; Wang, H.; Yin, Q. DNA Holliday Junction: History, Regulation and Bioactivity. Int. J. Mol. Sci. 2022, 23, 9730. [Google Scholar] [CrossRef]
- Mekonnen, N.; Yang, H.; Shin, Y.K. Homologous Recombination Deficiency in Ovarian, Breast, Colorectal, Pancreatic, Non-Small Cell Lung and Prostate Cancers, and the Mechanisms of Resistance to PARP Inhibitors. Front. Oncol. 2022, 12, 880643. [Google Scholar] [CrossRef]
- Shi, Z.; Chen, B.; Han, X.; Gu, W.; Liang, S.; Wu, L. Genomic and molecular landscape of homologous recombination deficiency across multiple cancer types. Sci. Rep. 2023, 13, 8899. [Google Scholar] [CrossRef]
- Shao, C.; Wan, J.; Lam, F.C.; Tang, H.; Marley, A.R.; Song, Y.; Miller, C.; Brown, M.; Han, J.; Adeboyeje, G. A comprehensive literature review and meta-analysis of the prevalence of pan-cancer BRCA mutations, homologous recombination repair gene mutations, and homologous recombination deficiencies. Environ. Mol. Mutagen. 2022, 63, 308. [Google Scholar] [CrossRef]
- Woodward, E.R.; Lalloo, F.; Forde, C.; Pugh, S.; Burghel, G.J.; Schlecht, H.; Harkness, E.F.; Howell, A.; Howell, S.J.; Gandhi, A.; et al. Germline testing of BRCA1, BRCA2, PALB2 and CHEK2 c.1100delC in 1514 triple negative familial and isolated breast cancers from a single centre, with extended testing of ATM, RAD51C and RAD51D in over 400. J. Med. Genet. 2023. [Google Scholar] [CrossRef]
- Shimelis, H.; LaDuca, H.; Hu, C.; Hart, S.N.; Na, J.; Thomas, A.; Akinhanmi, M.; Moore, R.M.; Brauch, H.; Cox, A.; et al. Triple-Negative Breast Cancer Risk Genes Identified by Multigene Hereditary Cancer Panel Testing. J. Natl. Cancer Inst. 2018, 110, 855. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Wang, S.M. The BRCAness Landscape of Cancer. Cells 2022, 11, 3877. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.J.; Ohsumi, S.; Takahashi, M.; Fukuma, E.; Jung, K.H.; Ishida, T.; Dai, M.S.; Chang, C.H.; Dalvi, T.; Walker, G.; et al. Prevalence of mutations in BRCA and homologous recombination repair genes and real-world standard of care of Asian patients with HER2-negative metastatic breast cancer starting first-line systemic cytotoxic chemotherapy: Subgroup analysis of the global BREAKOUT study. Breast Cancer 2022, 29, 92. [Google Scholar] [PubMed]
- Batalini, F.; Madison, R.W.; Sokol, E.S.; Jin, D.X.; Chen, K.T.; Decker, B.; Pavlick, D.C.; Frampton, G.M.; Wulf, G.M.; Garber, J.E.; et al. Homologous Recombination Deficiency Landscape of Breast Cancers and Real-World Effectiveness of Poly ADP-Ribose Polymerase Inhibitors in Patients with Somatic BRCA1/2, Germline PALB2, or Homologous Recombination Deficiency Signature. JCO Precis. Oncol. 2023, 7, e2300091. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Yu, X. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinform. 2016, 14, 131. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610. [Google Scholar] [CrossRef]
- Caron, M.C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagne, J.P.; Langelier, M.F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef]
- Basourakos, S.P.; Li, L.; Aparicio, A.M.; Corn, P.G.; Kim, J.; Thompson, T.C. Combination Platinum-based and DNA Damage Response-targeting Cancer Therapy: Evolution and Future Directions. Curr. Med. Chem. 2017, 24, 1586. [Google Scholar] [CrossRef]
- Creeden, J.F.; Nanavaty, N.S.; Einloth, K.R.; Gillman, C.E.; Stanbery, L.; Hamouda, D.M.; Dworkin, L.; Nemunaitis, J. Homologous recombination proficiency in ovarian and breast cancer patients. BMC Cancer 2021, 21, 1154. [Google Scholar] [CrossRef]
- Feng, Z.; Shao, D.; Cai, Y.; Bi, R.; Ju, X.; Chen, D.; Song, C.; Chen, X.; Li, J.; An, N.; et al. Homologous recombination deficiency status predicts response to platinum-based chemotherapy in Chinese patients with high-grade serous ovarian carcinoma. J. Ovarian Res. 2023, 16, 53. [Google Scholar] [CrossRef]
- Ferguson, L.R.; Chen, H.; Collins, A.R.; Connell, M.; Damia, G.; Dasgupta, S.; Malhotra, M.; Meeker, A.K.; Amedei, A.; Amin, A.; et al. Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin. Cancer Biol. 2015, 35, S5–S24. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, Y.H.; Lu, Q. Pro-oncogenic and anti-oncogenic pathways: Opportunities and challenges of cancer therapy. Future Oncol. 2010, 6, 587. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Frankum, J.R.; Punta, M.; Lise, S.; Alexander, J.; Chen, Y.; Yap, T.A.; Haider, S.; Tutt, A.N.J.; Lord, C.J. Clinical BRCA1/2 Reversion Analysis Identifies Hotspot Mutations and Predicted Neoantigens Associated with Therapy Resistance. Cancer Discov. 2020, 10, 1475. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Helenius, M.; Vaananen, K.; Bulanova, D.; Saarela, J.; Sokolenko, A.; Martens, J.; Imyanitov, E.; Kuznetsov, S. BRCA1-deficient breast cancer cell lines are resistant to MEK inhibitors and show distinct sensitivities to 6-thioguanine. Sci. Rep. 2016, 6, 28217. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef]
- Serio, P.; de Lima Pereira, G.F.; Katayama, M.L.H.; Roela, R.A.; Maistro, S.; Folgueira, M. Somatic Mutational Profile of High-Grade Serous Ovarian Carcinoma and Triple-Negative Breast Carcinoma in Young and Elderly Patients: Similarities and Divergences. Cells 2021, 10, 3586. [Google Scholar] [CrossRef]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef]
- Rempel, E.; Kluck, K.; Beck, S.; Ourailidis, I.; Kazdal, D.; Neumann, O.; Volckmar, A.L.; Kirchner, M.; Goldschmid, H.; Pfarr, N.; et al. Pan-cancer analysis of genomic scar patterns caused by homologous repair deficiency (HRD). NPJ Precis. Oncol. 2022, 6, 36. [Google Scholar] [CrossRef]
- Sharma, Y.; Miladi, M.; Dukare, S.; Boulay, K.; Caudron-Herger, M.; Gross, M.; Backofen, R.; Diederichs, S. A pan-cancer analysis of synonymous mutations. Nat. Commun. 2019, 10, 2569. [Google Scholar] [CrossRef]
- Zhang, X.; Sjoblom, T. Targeting Loss of Heterozygosity: A Novel Paradigm for Cancer Therapy. Pharmaceuticals 2021, 14, 57. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347. [Google Scholar] [CrossRef]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Reiniger, L.; Csabai, I.; Favero, F.; Birkbak, N.J.; Eklund, A.C.; Syed, A.; Szallasi, Z. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer 2018, 4, 16. [Google Scholar] [CrossRef]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274. [Google Scholar] [CrossRef] [PubMed]
- Fumet, J.D.; Lardenois, E.; Ray-Coquard, I.; Harter, P.; Joly, F.; Canzler, U.; Truntzer, C.; Tredan, O.; Liebrich, C.; Lortholary, A.; et al. Genomic Instability is Defined by Specific Tumor Microenvironment in Ovarian Cancer: A Subgroup Analysis of AGO OVAR 12 Trial. Cancers 2022, 14, 1189. [Google Scholar] [CrossRef] [PubMed]
- Lenz, L.; Neff, C.; Solimeno, C.; Cogan, E.S.; Abramson, V.G.; Boughey, J.C.; Falkson, C.; Goetz, M.P.; Ford, J.M.; Gradishar, W.J.; et al. Identifying homologous recombination deficiency in breast cancer: Genomic instability score distributions differ among breast cancer subtypes. Breast Cancer Res. Treat. 2023, 202, 191. [Google Scholar] [CrossRef] [PubMed]
- Wagener-Ryczek, S.; Merkelbach-Bruse, S.; Siemanowski, J. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Pers. Med. 2021, 11, 612. [Google Scholar] [CrossRef]
- Doig, K.D.; Fellowes, A.P.; Fox, S.B. Homologous Recombination Repair Deficiency: An Overview for Pathologists. Mod. Pathol. 2023, 36, 100049. [Google Scholar] [CrossRef]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454. [Google Scholar] [CrossRef] [PubMed]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef]
- Chiang, Y.C.; Lin, P.H.; Cheng, W.F. Homologous Recombination Deficiency Assays in Epithelial Ovarian Cancer: Current Status and Future Direction. Front. Oncol. 2021, 11, 675972. [Google Scholar] [CrossRef]
- Nagahashi, M.; Shimada, Y.; Ichikawa, H.; Kameyama, H.; Takabe, K.; Okuda, S.; Wakai, T. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci. 2019, 110, 6. [Google Scholar] [CrossRef]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776. [Google Scholar] [CrossRef]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366. [Google Scholar] [CrossRef] [PubMed]
- Blanc-Durand, F.; Yaniz-Galende, E.; Llop-Guevara, A.; Genestie, C.; Serra, V.; Herencia-Ropero, A.; Klein, C.; Berton, D.; Lortholary, A.; Dohollou, N.; et al. A RAD51 functional assay as a candidate test for homologous recombination deficiency in ovarian cancer. Gynecol. Oncol. 2023, 171, 106. [Google Scholar] [CrossRef] [PubMed]
- Tumiati, M.; Hietanen, S.; Hynninen, J.; Pietila, E.; Farkkila, A.; Kaipio, K.; Roering, P.; Huhtinen, K.; Alkodsi, A.; Li, Y.; et al. A Functional Homologous Recombination Assay Predicts Primary Chemotherapy Response and Long-Term Survival in Ovarian Cancer Patients. Clin. Cancer Res. 2018, 24, 4482. [Google Scholar] [CrossRef]
- Graeser, M.; McCarthy, A.; Lord, C.J.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.S.; et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin. Cancer Res. 2010, 16, 6159. [Google Scholar] [CrossRef] [PubMed]
- Ladan, M.M.; van Gent, D.C.; Jager, A. Homologous Recombination Deficiency Testing for BRCA-Like Tumors: The Road to Clinical Validation. Cancers 2021, 13, 1004. [Google Scholar] [CrossRef]
- Stewart, M.D.; Merino Vega, D.; Arend, R.C.; Baden, J.F.; Barbash, O.; Beaubier, N.; Collins, G.; French, T.; Ghahramani, N.; Hinson, P.; et al. Homologous Recombination Deficiency: Concepts, Definitions, and Assays. Oncologist 2022, 27, 167. [Google Scholar] [CrossRef]
- Hong, J.; Lee, J.; Kwon, M.; Kim, J.Y.; Kim, J.W.; Ahn, J.S.; Im, Y.H.; Park, Y.H. Local Laboratory Testing of Germline BRCA Mutations vs. Myriad: A Single-Institution Experience in Korea. Diagnostics 2021, 11, 370. [Google Scholar] [CrossRef]
- Gruber, J.J.; Afghahi, A.; Timms, K.; DeWees, A.; Gross, W.; Aushev, V.N.; Wu, H.T.; Balcioglu, M.; Sethi, H.; Scott, D.; et al. A phase II study of talazoparib monotherapy in patients with wild-type BRCA1 and BRCA2 with a mutation in other homologous recombination genes. Nat. Cancer 2022, 3, 1181. [Google Scholar] [CrossRef]
- Milbury, C.A.; Creeden, J.; Yip, W.K.; Smith, D.L.; Pattani, V.; Maxwell, K.; Sawchyn, B.; Gjoerup, O.; Meng, W.; Skoletsky, J.; et al. Clinical and analytical validation of FoundationOne(R)CDx, a comprehensive genomic profiling assay for solid tumors. PLoS ONE 2022, 17, e0264138. [Google Scholar] [CrossRef]
- Menezes, M.C.S.; Raheem, F.; Mina, L.; Ernst, B.; Batalini, F. PARP Inhibitors for Breast Cancer: Germline BRCA1/2 and Beyond. Cancers 2022, 14, 4332. [Google Scholar] [CrossRef]
- Mirza, M.R.; Lindahl, G.; Mahner, S.; Redondo, A.; Fabbro, M.; Rimel, B.J.; Herrstedt, J.; Oza, A.M.; Canzler, U.; Berek, J.S.; et al. Ad hoc Analysis of the Phase III ENGOT-OV16/NOVA Study: Niraparib Efficacy in Germline BRCA Wild-type Recurrent Ovarian Cancer with Homologous Recombination Repair Defects. Cancer Res. Commun. 2022, 2, 1436. [Google Scholar] [CrossRef]
- Batalini, F.; Gulhan, D.C.; Mao, V.; Tran, A.; Polak, M.; Xiong, N.; Tayob, N.; Tung, N.M.; Winer, E.P.; Mayer, E.L.; et al. Mutational Signature 3 Detected from Clinical Panel Sequencing is Associated with Responses to Olaparib in Breast and Ovarian Cancers. Clin. Cancer Res. 2022, 28, 4714. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol. 2017, 28, 512. [Google Scholar] [CrossRef]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. 2021, 32, 240. [Google Scholar] [CrossRef]
- Finotello, F.; Trajanoski, Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol. Immunother. 2018, 67, 1031. [Google Scholar] [CrossRef]
- Ying-Rui, M.; Bu-Fan, B.; Deng, L.; Rong, S.; Qian-Mei, Z. Targeting the stimulator of interferon genes (STING) in breast cancer. Front. Pharmacol. 2023, 14, 1199152. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhuang, Z.; Li, J.; Feng, Z. Significance of the cGAS-STING Pathway in Health and Disease. Int. J. Mol. Sci. 2023, 24, 13316. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Liu, D.; Wang, X.; Guo, Z.; Sun, H.; Song, Y.; Wang, D. DNA Damage and Activation of cGAS/STING Pathway Induce Tumor Microenvironment Remodeling. Front. Cell Dev. Biol. 2021, 9, 828657. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Kim, S.Y.; Kim, S.H.; Choi, S.; Lee, B.; Shin, J.S. STING mediates nuclear PD-L1 targeting-induced senescence in cancer cells. Cell Death Dis. 2022, 13, 791. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, B.; Musolino, A.; Llop-Guevara, A.; Serra, V.; De Silva, P.; Hlavata, Z.; Sangiolo, D.; Willard-Gallo, K.; Solinas, C. Homologous Recombination Repair Deficiency and the Immune Response in Breast Cancer: A Literature Review. Transl. Oncol. 2020, 13, 410. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108. [Google Scholar] [CrossRef]
- Laskowski, T.J.; Biederstadt, A.; Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 2022, 22, 557. [Google Scholar] [CrossRef]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453. [Google Scholar] [CrossRef]
- Siemaszko, J.; Marzec-Przyszlak, A.; Bogunia-Kubik, K. NKG2D Natural Killer Cell Receptor-A Short Description and Potential Clinical Applications. Cells 2021, 10, 1420. [Google Scholar] [CrossRef]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The Natural Cytotoxicity Receptors in Health and Disease. Front. Immunol. 2019, 10, 909. [Google Scholar] [CrossRef] [PubMed]
- Gascon-Ruiz, M.; Ramirez-Labrada, A.; Lastra, R.; Martinez-Lostao, L.; Pano-Pardo, J.R.; Sesma, A.; Zapata-Garcia, M.; Moratiel, A.; Quilez, E.; Torres-Ramon, I.; et al. A Subset of PD-1-Expressing CD56(bright) NK Cells Identifies Patients with Good Response to Immune Checkpoint Inhibitors in Lung Cancer. Cancers 2023, 15, 329. [Google Scholar] [CrossRef]
- Lu, L.; Yang, C.; Zhou, X.; Wu, L.; Hong, X.; Li, W.; Wang, X.; Yang, Y.; Cao, D.; Zhang, A.; et al. STING signaling promotes NK cell antitumor immunity and maintains a reservoir of TCF-1(+) NK cells. Cell Rep. 2023, 42, 113108. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef]
- Allison, E.; Edirimanne, S.; Matthews, J.; Fuller, S.J. Breast Cancer Survival Outcomes and Tumor-Associated Macrophage Markers: A Systematic Review and Meta-Analysis. Oncol. Ther. 2023, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Kotsifaki, A.; Alevizopoulos, N.; Dimopoulou, V.; Armakolas, A. Unveiling the Immune Microenvironment’s Role in Breast Cancer: A Glimpse into Promising Frontiers. Int. J. Mol. Sci. 2023, 24, 15332. [Google Scholar] [CrossRef]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860. [Google Scholar] [CrossRef]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44. [Google Scholar] [CrossRef]
- Emens, L.A.; Adams, S.; Barrios, C.H.; Dieras, V.; Iwata, H.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 2021, 32, 983. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.G.; Kim, S.K.; Shepherd, J.H.; Cha, Y.J.; Bae, S.J.; Kim, C.; Jeong, J.; Perou, C.M. Clinical and genomic assessment of PD-L1 SP142 expression in triple-negative breast cancer. Breast Cancer Res. Treat. 2021, 188, 165. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810. [Google Scholar] [CrossRef] [PubMed]
- Winer, E.P.; Lipatov, O.; Im, S.A.; Goncalves, A.; Munoz-Couselo, E.; Lee, K.S.; Schmid, P.; Tamura, K.; Testa, L.; Witzel, I.; et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 499. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47(+)/CD73(+)/PDL1(+) immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509. [Google Scholar] [CrossRef]
- Wang, H.Y.; Deng, L.; Li, Y.Q.; Zhang, X.; Long, Y.K.; Zhang, X.; Feng, Y.F.; He, Y.; Tang, T.; Yang, X.H.; et al. Pan-cancer Analysis of Tumor Mutational Burden and Homologous Recombination DNA Damage Repair Using Targeted Next-Generation Sequencing. Cancer Res. Treat. 2021, 53, 973. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients with Previously Untreated BRAF Wild-Type Advanced Melanoma Treated with Nivolumab Therapy: Three-Year Follow-up of a Randomized Phase 3 Trial. JAMA Oncol. 2019, 5, 187. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Rutkowski, P.; Hassel, J.C.; McNeil, C.M.; Kalinka, E.A.; et al. Five-Year Outcomes with Nivolumab in Patients with Wild-Type BRAF Advanced Melanoma. J. Clin. Oncol. 2020, 38, 3937. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Lv, M.; He, X.; Pan, Y.; Ge, J.; Hu, X.; Zheng, Y.; Huang, M.; Zhou, C.; You, C. Homologous recombination repair gene mutations as a predictive biomarker for immunotherapy in patients with advanced melanoma. Front. Immunol. 2022, 13, 871756. [Google Scholar] [CrossRef] [PubMed]
- Montisci, A.; Vietri, M.T.; Palmieri, V.; Sala, S.; Donatelli, F.; Napoli, C. Cardiac Toxicity Associated with Cancer Immunotherapy and Biological Drugs. Cancers 2021, 13, 4797. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; Meylan, M.; de Reynies, A.; Sautes-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409. [Google Scholar] [CrossRef]
- Cortellini, A.; Bersanelli, M.; Buti, S.; Gambale, E.; Atzori, F.; Zoratto, F.; Parisi, A.; Brocco, D.; Pireddu, A.; Cannita, K.; et al. Family history of cancer as surrogate predictor for immunotherapy with anti-PD1/PD-L1 agents: Preliminary report of the FAMI-L1 study. Immunotherapy 2018, 10, 643. [Google Scholar] [CrossRef]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329. [Google Scholar] [CrossRef]
- Peshkin, B.N.; Alabek, M.L.; Isaacs, C. BRCA1/2 mutations and triple negative breast cancers. Breast Dis. 2010, 32, 25. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Jain, E.; Cohen, O.; Kim, D.; Buendia-Buendia, J.; Winer, E.; Lin, N.; Tolaney, S.M.; Wagle, N. Prevalence and mutational determinants of high tumor mutation burden in breast cancer. Ann. Oncol. 2020, 31, 387. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients with Breast Cancer. J. Clin. Oncol. 2016, 34, 1460. [Google Scholar] [CrossRef]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304. [Google Scholar] [CrossRef]
- Symmans, W.F.; Wei, C.; Gould, R.; Yu, X.; Zhang, Y.; Liu, M.; Walls, A.; Bousamra, A.; Ramineni, M.; Sinn, B.; et al. Long-Term Prognostic Risk after Neoadjuvant Chemotherapy Associated with Residual Cancer Burden and Breast Cancer Subtype. J. Clin. Oncol. 2017, 35, 1049. [Google Scholar] [CrossRef]
- Alba, E.; Calvo, L.; Albanell, J.; De la Haba, J.R.; Arcusa Lanza, A.; Chacon, J.I.; Sanchez-Rovira, P.; Plazaola, A.; Lopez Garcia-Asenjo, J.A.; Bermejo, B.; et al. Chemotherapy (CT) and hormonotherapy (HT) as neoadjuvant treatment in luminal breast cancer patients: Results from the GEICAM/2006-03, a multicenter, randomized, phase-II study. Ann. Oncol. 2012, 23, 3069. [Google Scholar] [CrossRef]
- Shepherd, J.H.; Ballman, K.; Polley, M.C.; Campbell, J.D.; Fan, C.; Selitsky, S.; Fernandez-Martinez, A.; Parker, J.S.; Hoadley, K.A.; Hu, Z.; et al. CALGB 40603 (Alliance): Long-Term Outcomes and Genomic Correlates of Response and Survival after Neoadjuvant Chemotherapy with or without Carboplatin and Bevacizumab in Triple-Negative Breast Cancer. J. Clin. Oncol. 2022, 40, 1323. [Google Scholar] [CrossRef]
- Byrski, T.; Huzarski, T.; Dent, R.; Marczyk, E.; Jasiowka, M.; Gronwald, J.; Jakubowicz, J.; Cybulski, C.; Wisniowski, R.; Godlewski, D.; et al. Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res. Treat. 2014, 147, 401. [Google Scholar] [CrossRef]
- Loibl, S.; Weber, K.E.; Timms, K.M.; Elkin, E.P.; Hahnen, E.; Fasching, P.A.; Lederer, B.; Denkert, C.; Schneeweiss, A.; Braun, S.; et al. Survival analysis of carboplatin added to an anthracycline/taxane-based neoadjuvant chemotherapy and HRD score as predictor of response-final results from GeparSixto. Ann. Oncol. 2018, 29, 2341. [Google Scholar] [CrossRef]
- Geyer, C.E.; Sikov, W.M.; Huober, J.; Rugo, H.S.; Wolmark, N.; O’Shaughnessy, J.; Maag, D.; Untch, M.; Golshan, M.; Lorenzo, J.P.; et al. Long-term efficacy and safety of addition of carboplatin with or without veliparib to standard neoadjuvant chemotherapy in triple-negative breast cancer: 4-year follow-up data from BrighTNess, a randomized phase III trial. Ann. Oncol. 2022, 33, 384. [Google Scholar] [CrossRef]
- Tung, N.; Arun, B.; Hacker, M.R.; Hofstatter, E.; Toppmeyer, D.L.; Isakoff, S.J.; Borges, V.; Legare, R.D.; Isaacs, C.; Wolff, A.C.; et al. TBCRC 031: Randomized Phase II Study of Neoadjuvant Cisplatin Versus Doxorubicin-Cyclophosphamide in Germline BRCA Carriers with HER2-Negative Breast Cancer (the INFORM trial). J. Clin. Oncol. 2020, 38, 1539. [Google Scholar] [CrossRef]
- Geyer, C.E., Jr.; Garber, J.E.; Gelber, R.D.; Yothers, G.; Taboada, M.; Ross, L.; Rastogi, P.; Cui, K.; Arahmani, A.; Aktan, G.; et al. Overall survival in the OlympiA phase III trial of adjuvant olaparib in patients with germline pathogenic variants in BRCA1/2 and high-risk, early breast cancer. Ann. Oncol. 2022, 33, 1250. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517. [Google Scholar] [CrossRef]
- Incorvaia, L.; Perez, A.; Marchetti, C.; Brando, C.; Gristina, V.; Cancelliere, D.; Pivetti, A.; Contino, S.; Di Giovanni, E.; Barraco, N.; et al. Theranostic biomarkers and PARP-inhibitors effectiveness in patients with non-BRCA associated homologous recombination deficient tumors: Still looking through a dirty glass window? Cancer Treat. Rev. 2023, 121, 102650. [Google Scholar] [CrossRef]
- Mayer, E.L.; Abramson, V.; Jankowitz, R.; Falkson, C.; Marcom, P.K.; Traina, T.; Carey, L.; Rimawi, M.; Specht, J.; Miller, K.; et al. TBCRC 030: A phase II study of preoperative cisplatin versus paclitaxel in triple-negative breast cancer: Evaluating the homologous recombination deficiency (HRD) biomarker. Ann. Oncol. 2020, 31, 1518. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628. [Google Scholar] [CrossRef]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Telli, M.L.; Rugo, H.S.; Mailliez, A.; Ettl, J.; Grischke, E.M.; Mina, L.A.; Balmana, J.; Fasching, P.A.; Hurvitz, S.A.; et al. A Phase II Study of Talazoparib after Platinum or Cytotoxic Nonplatinum Regimens in Patients with Advanced Breast Cancer and Germline BRCA1/2 Mutations (ABRAZO). Clin. Cancer Res. 2019, 25, 2717. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753. [Google Scholar] [CrossRef]
- Dieras, V.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Veliparib with carboplatin and paclitaxel in BRCA-mutated advanced breast cancer (BROCADE3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1269. [Google Scholar] [CrossRef]
- Tovey, H.; Sipos, O.; Parker, J.S.; Hoadley, K.A.; Quist, J.; Kernaghan, S.; Kilburn, L.; Salgado, R.; Loi, S.; Kennedy, R.D.; et al. Integrated Multimodal Analyses of DNA Damage Response and Immune Markers as Predictors of Response in Metastatic Triple-Negative Breast Cancer in the TNT Trial (NCT00532727). Clin. Cancer Res. 2023, 29, 3691. [Google Scholar] [CrossRef]
- Zhao, E.Y.; Shen, Y.; Pleasance, E.; Kasaian, K.; Leelakumari, S.; Jones, M.; Bose, P.; Ch’ng, C.; Reisle, C.; Eirew, P.; et al. Homologous Recombination Deficiency and Platinum-Based Therapy Outcomes in Advanced Breast Cancer. Clin. Cancer Res. 2017, 23, 7521. [Google Scholar] [CrossRef]
- Quan, M.L.; Olivotto, I.A.; Baxter, N.N.; Friedenreich, C.M.; Metcalfe, K.; Warner, E.; MacLennan, K.; Stephen, J.E.; Akbari, M.R.; Howell, D.; et al. A pan-Canadian prospective study of young women with breast cancer: The rationale and protocol design for the RUBY study. Curr. Oncol. 2020, 27, e516. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560. [Google Scholar] [CrossRef]
- Mao, Y.; Qu, Q.; Chen, X.; Huang, O.; Wu, J.; Shen, K. The Prognostic Value of Tumor-Infiltrating Lymphocytes in Breast Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0152500. [Google Scholar] [CrossRef]
- Teo, M.Y.; Seier, K.; Ostrovnaya, I.; Regazzi, A.M.; Kania, B.E.; Moran, M.M.; Cipolla, C.K.; Bluth, M.J.; Chaim, J.; Al-Ahmadie, H.; et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J. Clin. Oncol. 2018, 36, 1685. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intervention | Identifier# | Title | HRD Score | Status |
|---|---|---|---|---|
| Carboplatin plus background treatment | NCT01426880 | Addition of Carboplatin to Neoadjuvant Therapy for Triple-negative and HER2-positive Early Breast Cancer | HRD ≥ 42: 70.5% tBRCA1/2mt: 29% gBRCA1/2mt: 20% | Completed |
| Cisplatin | NCT01630226 | Cisplatin Monotherapy in the Treatment of BRCA1 Positive Breast Cancer Patients in Poland | gBRCA1/2mt: 100% | Unknown |
| Carboplatin + nanoparticle albumin-bound paclitaxel + vorinostat (HDAC inhibitor) | NCT00616967 | Carboplatin and Nab-Paclitaxel with or without Vorinostat in Treating Women with Newly Diagnosed Operable Breast Cancer | HRD ≥ 42: 46% | Active; not recruiting |
| Carboplatin + Eribulin | NCT01372579 | Carboplatin and Eribulin Mesylate in Triple-negative Breast Cancer Patients | HRD ≥ 42: 46% gBRCA1/2mt: 10% | Unknown |
| Cisplatin vs. Doxorubicin/Cyclophosphamide | NCT01670500 | Cisplatin vs. Doxorubicin/Cyclophosphamide in BrCa | gBRCA1/2mt: 68% | Active; not recruiting |
| Cisplatin vs. Paclitaxel | NCT01982448 | Cisplatin vs. Paclitaxel for Triple-negative Breast Cancer | HRD ≥ 33: 71% | Completed |
| Carboplatin + gemcitabine + iniparib | NCT00813956 | A Phase 2 Study of Standard Chemotherapy Plus BSI-201 (a PARP Inhibitor) in the Neoadjuvant Treatment of Triple-negative Breast Cancer | gBRCA1/2mt: 24% | Completed |
| Paclitaxel vs. paclitaxel + veliparib + carboplatin | NCT01042379 | I-SPY TRIAL: Neoadjuvant and Personalized Adaptive Novel Agents to Treat Breast Cancer | gBRCA1/2mt: 17% | Actively recruiting |
| Paclitaxel vs. paclitaxel + carboplatin vs. paclitaxel + carboplatin + veliparib | NCT02032277 | A Study Evaluating Safety and Efficacy of the Addition of ABT-888 Plus Carboplatin Versus the Addition of Carboplatin to Standard Chemotherapy Versus Standard Chemotherapy in Subjects with Early-Stage Triple-negative Breast Cancer | HRD ≥ 42: 67% gBRCA1/2mt: 15% | Completed |
| Paclitaxel-olaparib vs. paclitaxel-carboplatin | NCT02789332 | Assessing the Efficacy of Paclitaxel and Olaparib in Comparison to Paclitaxel/Carboplatin Followed by Epirubicin/Cyclophosphamide as Neoadjuvant Chemotherapy in Patients with HER2-negative Early Breast Cancer and Homologous Recombination Deficiency | g/tBRCA1/2mt: 56.2% | Completed |
| Talazoparib | NCT03499353 | Talazoparib For Neoadjuvant Treatment of Germline BRCA1/2 Mutation Patients with Early Human Epidermal-Growth-Factor-Receptor-2-Negative Breast Cancer | gBRCA1mt: 42.1% gBRCA2mt: 10.5% | Terminated (Not due to safety concerns) |
| Talazoparib | NCT02282345 | Talazoparib Before Standard Therapy in Treating Patients with Invasive, BRCA-Mutated Breast Cancer | gBRCA1mt: 78.8% gBRCA2mt: 21.2% | Completed |
| Olaparib | NCT02624973 | Personalized Treatment of High-risk Mammary Cancer—the PETREMAC Trial | HRD: 34% gBRCA1/2mt: 14% | Active; not recruiting |
| Niraparib | NCT03329937 | Study Evaluating the Antitumor Activity and Safety of Niraparib as Neoadjuvant Treatment in Participants with Breast Cancer | gBRCA1mt: 67% gBRCA2mt: 28% | Completed |
| Olaparib | NCT02032823 | Olaparib as Adjuvant Treatment in Patients with Germline BRCA Mutated High-Risk HER2 Negative Primary Breast Cancer | gBRCA1mt: 72% gBRCA2mt: 27% | Active; not recruiting |
| Rucaparib | EudraCT 2014-003319-12 | Window study of the PARP inhibitor rucaparib in patients with primary triple-negative or BRCA1/2-related breast cancer | HRD: 69% gBRCA1/2mt: 19% | Completed |
| Intervention | Identifier# | Title | HRD Score | Status |
|---|---|---|---|---|
| Cisplatin or carboplatin | NCT00483223 | Platinum for Triple-negative Metastatic Breast Cancer and Evaluation of p63/p73 as a Biomarker of Response | HRD score (LOH: 12.68; LST: 5.11) | Completed |
| Carboplatin vs. Docetaxel | NCT00532727 | Triple-negative Breast Cancer Trial (TNT) | gBRCA1/2 mt BRCA1 methylation HRD score ≥ 42 | Unknown |
| Cisplatin | H14-00681-A019 | Homologous Recombination Deficiency and Platinum-Based Therapy Outcomes in Advanced Breast Cancer | HRD score(WGS) 20% | Completed |
| Carboplatin or Cisplatin | N/A | Efficacy of platinum-based chemotherapy in metastatic breast cancer and HRD biomarkers: utility of exome sequencing | HRD score and COSMIC signature 3 (WES) | Completed |
| Carboplatin + Paclitaxel vs. Carboplatin + Paclitaxel + veliparib vs. Veliparib | NCT02163694 | A Phase 3 Randomized, Placebo-controlled Trial of Carboplatin and Paclitaxel with or without Veliparib (ABT-888) in HER2-negative Metastatic or Locally Advanced Unresectable BRCA-associated Breast Cancer | gBRCA1/2 mt | Active; not recruiting |
| Olaparib | NCT02000622 | Assessment of the Efficacy and Safety of Olaparib Monotherapy Versus Physicians’ Choice Chemotherapy in the Treatment of Metastatic Breast Cancer Patients with Germline BRCA1/2 Mutations | gBRCA1/2 mt | Active; not recruiting |
| Talazoparib | NCT01945775 | A Study Evaluating Talazoparib (BMN 673), a PARP Inhibitor, in Advanced and/or Metastatic Breast Cancer Patients with BRCA Mutation | gBRCA1/2 mt | Completed |
| Niraparib + Pembrolizumab | NCT02657889 | Niraparib in Combination with Pembrolizumab in Patients with Triple-negative Breast Cancer or Ovarian Cancer | gBRCA1/2 mt | Completed |
| Olaparib + durvalumab | NCT02734004 | A Phase I/II Study of MEDI4736 in Combination with Olaparib in Patients with Advanced Solid Tumors | gBRCA1/2 mt | Active; not recruiting |
| Olaparib | NCT03344965 | Olaparib In Metastatic Breast Cancer | HRR pathway gene mutations | Recruiting |
| Rucaparib | NCT02505048 | A Study to Assess the Efficacy of Rucaparib in Metastatic Breast Cancer Patients with a BRCAness Genomic Signature | LOH score or HRR pathway gene mutations | Completed |
| Talazoparib | NCT02401347 | Phase II Trial of Talazoparib in BRCA1/2 Wild-type HER2-negative Breast Cancer and Other Solid Tumors | BRCA WT with HRR pathway gene mutations | Completed |
| Palbociclib + Olaparib + Fulvestrant | NCT03685331 | HOPE: Olaparib, Palbociclib, and Fulvestrant in Patients with BRCA-Mutation-associated, HR+, HER2-metastatic Breast Cancer | gBRCA1/2 mt | Active; not recruiting |
| Durvalumab + Olaparib + Fulvestrant | NCT04053322 | Durvalumab, with Olaparib and Fulvestrant in Advanced ER+, HER2- Breast Cancer Patients | gBRCA1/2 mt and HRR pathway gene mutations | Active; not recruiting |
| Pembrolizumab + Olaparib | NCT03025035 | Pembrolizumab in Combination with Olaparib in Advanced BRCA-mutated or HDR-defect Breast Cancer | gBRCA1/2 mt and HRR pathway gene mutations | Recruiting |
| Niraparib, anti-TIM3, bevacizumab, and platinum-based doublet chemotherapy + anti-PD-1 | NCT03307785 | Study of Niraparib, TSR-022, Bevacizumab, and Platinum-Based Doublet Chemotherapy in Combination with TSR-042 | HRD not a criterion | Active; not recruiting |
| Talazoparib + avelumab | NCT03565991 | Javelin BRCA/ATM: Avelumab Plus Talazoparib in Patients with BRCA or ATM Mutant Solid Tumors | BRCA or ATM mutations | Not Recruiting (study continued as NCT05059522) |
| Rucaparib + atezolizumab | NCT03101280 | A Combination Study of Rucaparib and Atezolizumab in Participants with Advanced Gynecologic Cancers and Triple-negative Breast Cancer | LOH or HRR pathway gene mutations | Completed |
| Olaparib + durvalumab | NCT02484404 | Phase I/II Study of the Anti-Programmed Death Ligand-1 Durvalumab Antibody (MEDI4736) in Combination with Olaparib and/or Cediranib for Advanced Solid Tumors and Advanced or Recurrent Ovarian, Triple-negative Breast, Lung, Prostate, and Colorectal Cancers | gBRCA1/2 mt | Recruiting |
| Olaparib + durvalumab + copanlisib | NCT03842228 | Testing the Combination of the Anti-cancer Drugs Copanlisib, Olaparib, and MEDI4736 (Durvalumab) in Patients with Advanced Solid Tumors with Selected Mutations | HRR pathway gene mutations | Recruiting |
| Olaparib + durvalumab | NCT03167619 | Phase II Multicenter Study of Durvalumab and Olaparib in Platinum tReated Advanced Triple-negative Breast Cancer (DORA) | HRD not a criterion | Completed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, U.; Vungarala, S.; Tiriveedhi, V. Genomic Features of Homologous Recombination Deficiency in Breast Cancer: Impact on Testing and Immunotherapy. Genes 2024, 15, 162. https://doi.org/10.3390/genes15020162
Ali U, Vungarala S, Tiriveedhi V. Genomic Features of Homologous Recombination Deficiency in Breast Cancer: Impact on Testing and Immunotherapy. Genes. 2024; 15(2):162. https://doi.org/10.3390/genes15020162
Chicago/Turabian StyleAli, Umer, Sunitha Vungarala, and Venkataswarup Tiriveedhi. 2024. "Genomic Features of Homologous Recombination Deficiency in Breast Cancer: Impact on Testing and Immunotherapy" Genes 15, no. 2: 162. https://doi.org/10.3390/genes15020162