The Genetic Basis of the First Patient with Wiedemann–Rautenstrauch Syndrome in the Russian Federation

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Subject and Methods
2.1. Clinical Evaluation
2.2. Molecular Tests
3. Results
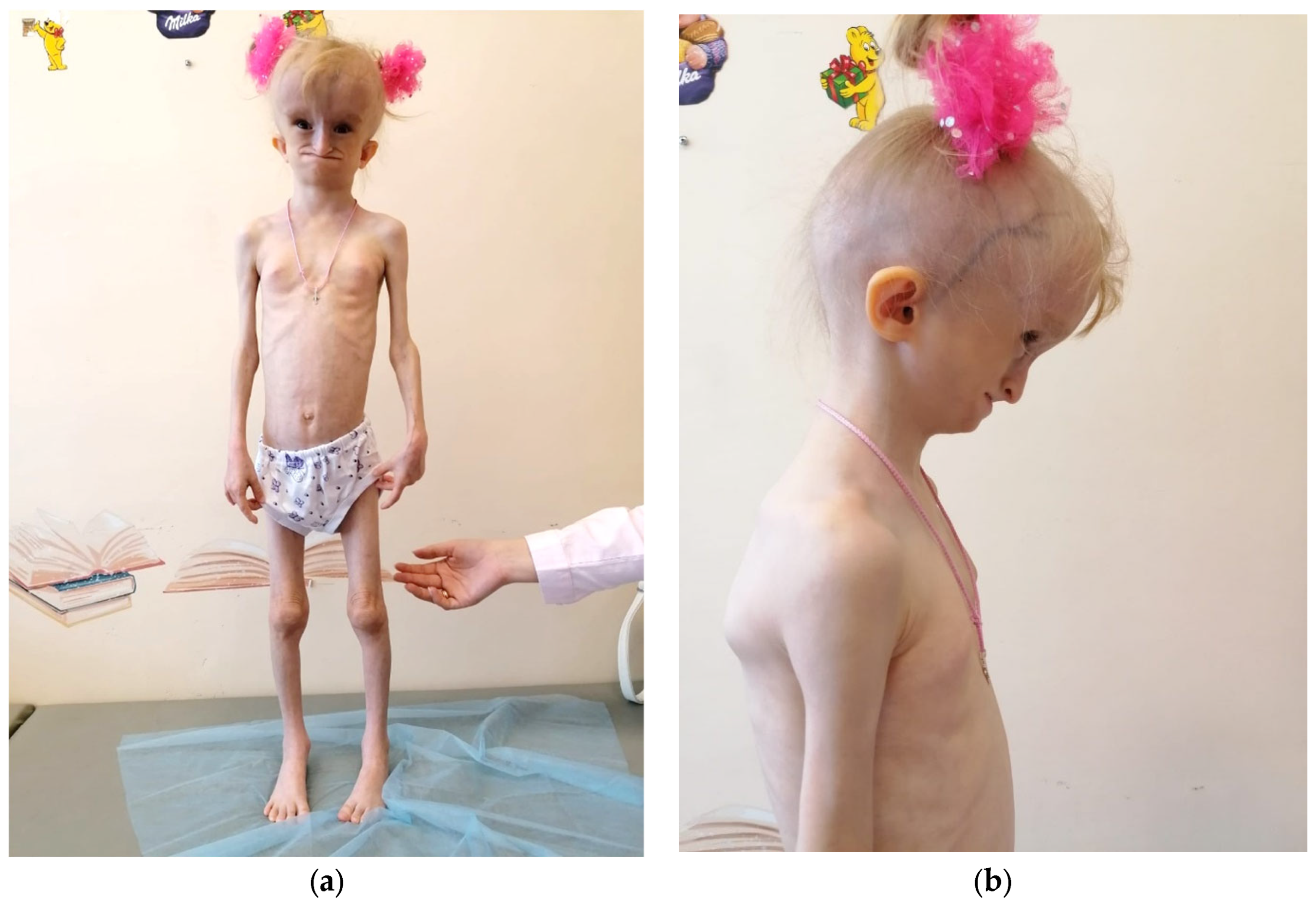
3.1. Clinical Findings
3.2. Molecular Tests Results
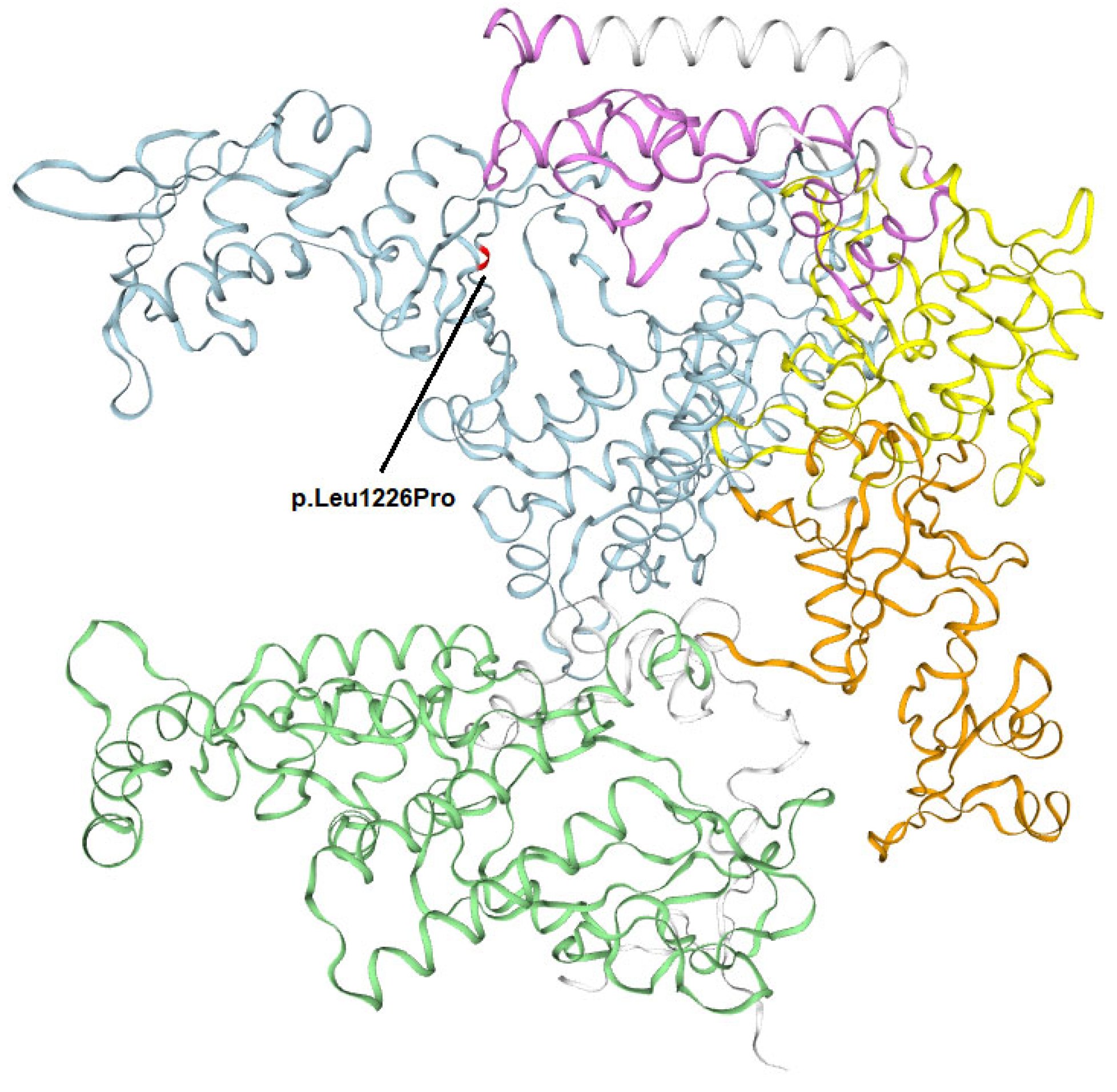
3.2.1. Whole-Exome Sequencing (WES)
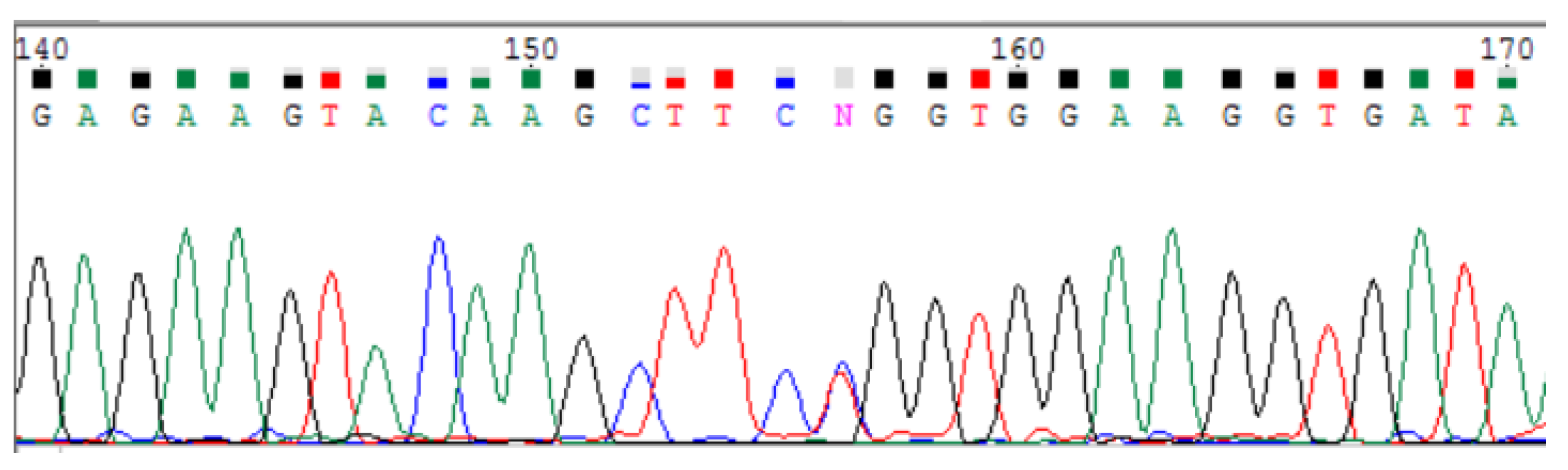
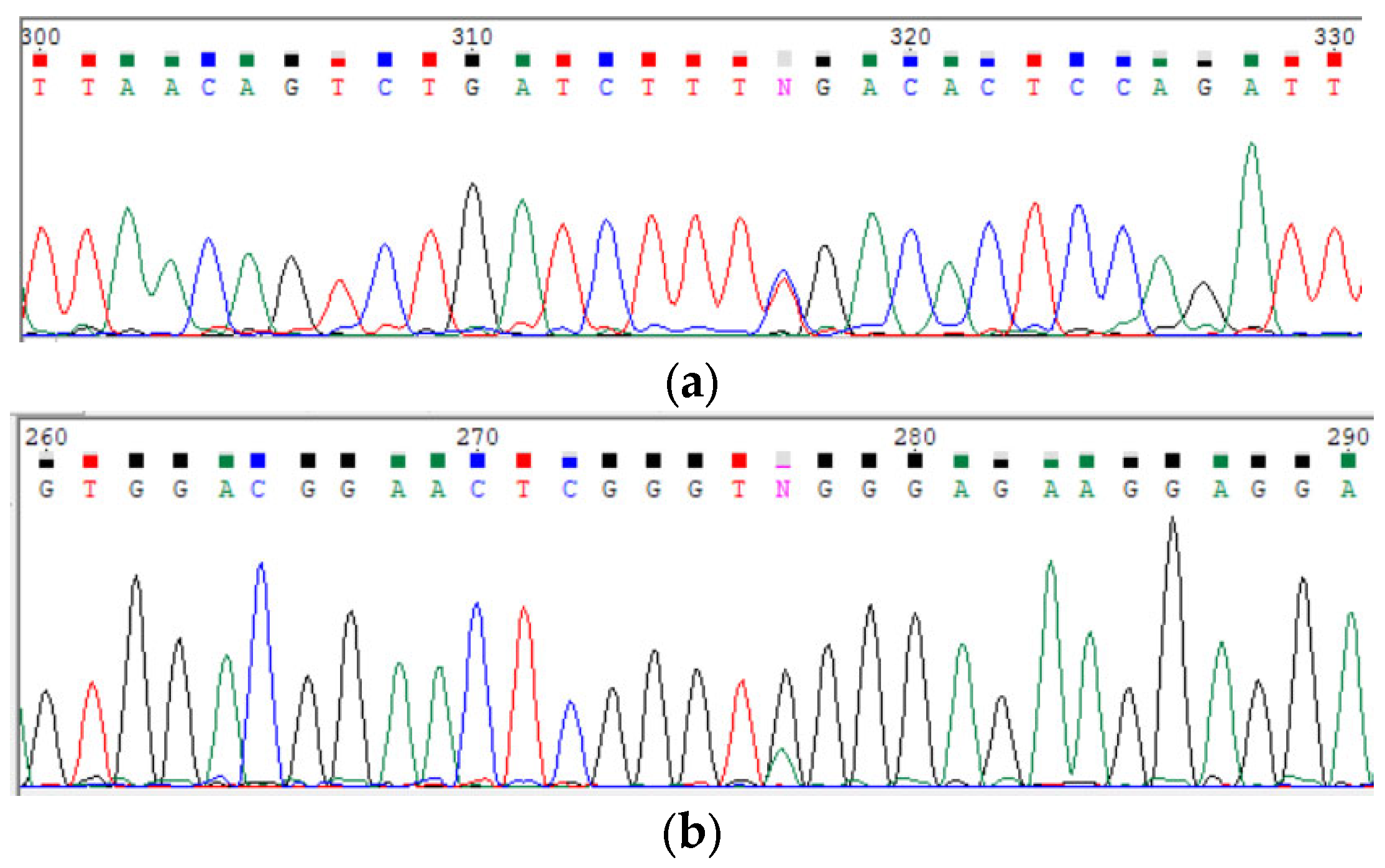
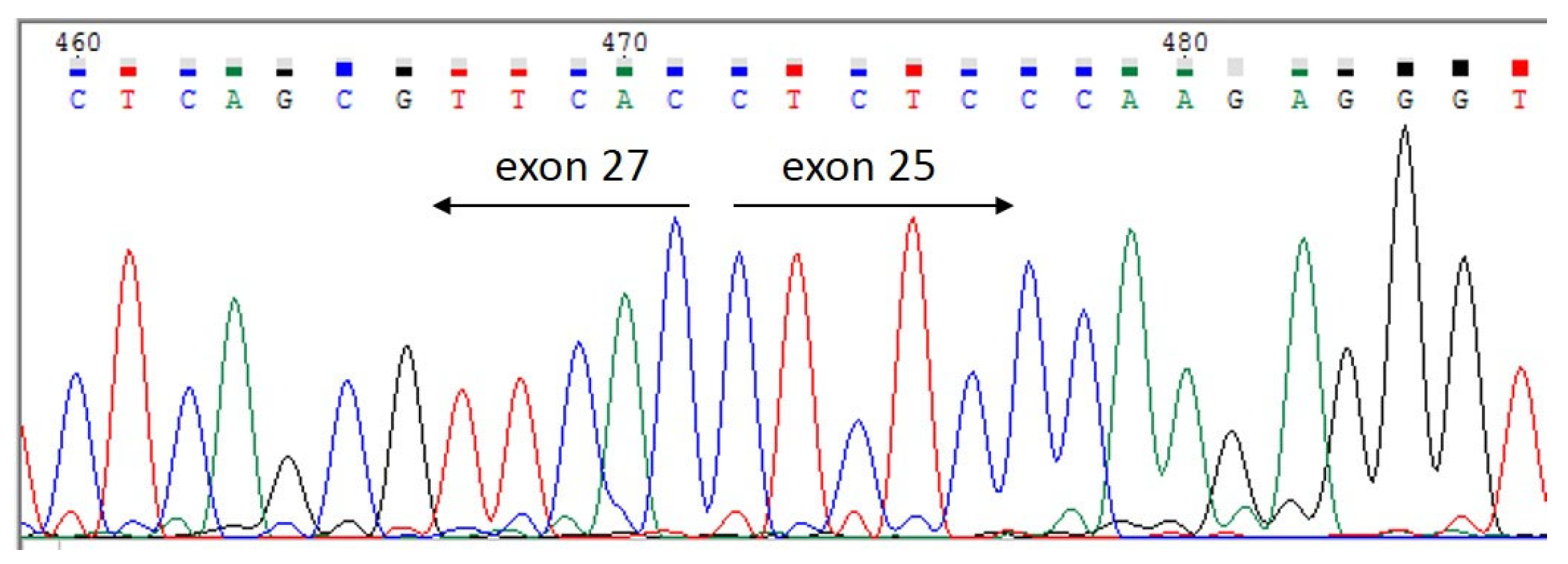
3.2.2. Sanger Sequencing
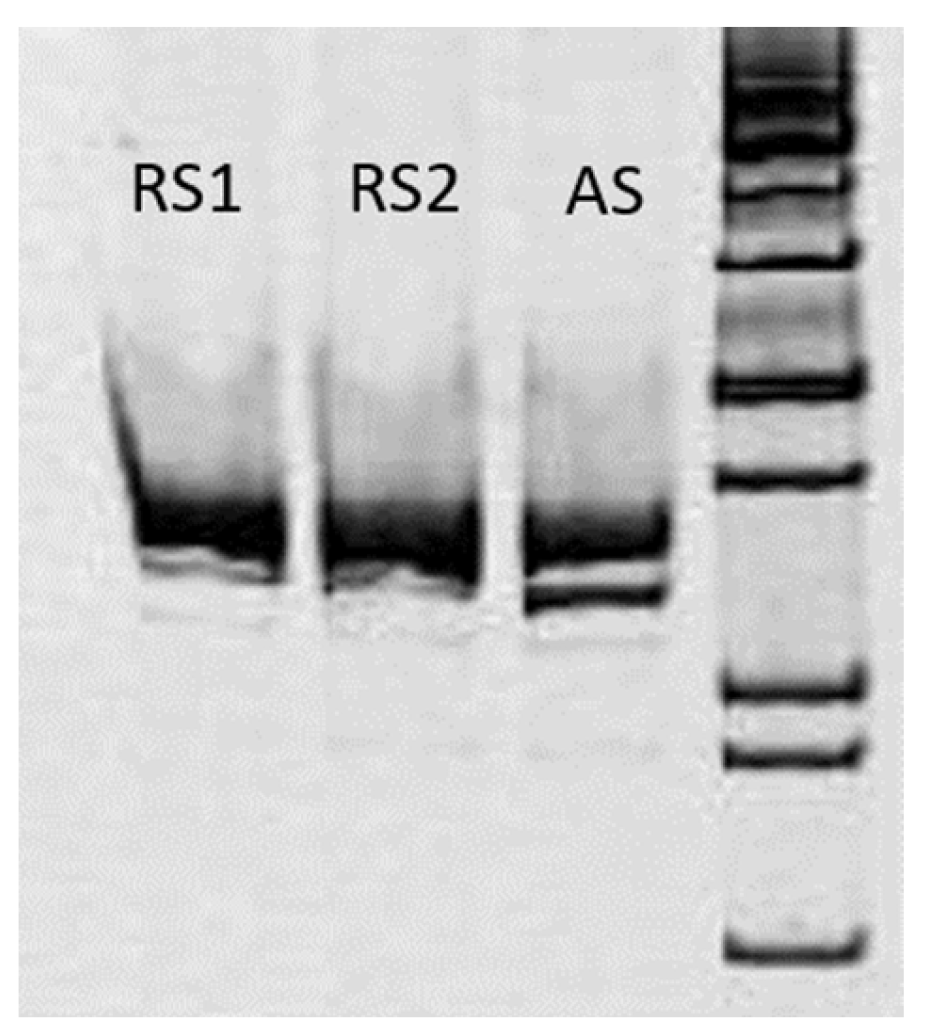
3.2.3. mRNA Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Infante, J.; Serrano-Cárdenas, K.M.; Corral-Juan, M.; Farré, X.; Sánchez, I.; de Lucas, E.M.; García, A.; Martín-Gurpegui, J.L.; Berciano, J.; Matilla-Dueñas, A. POLR3A-related spastic ataxia: New mutations and a look into the phenotype. J. Neurol. 2020, 267, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Daoud, H.; Tétreault, M.; Gibson, W.; Guerrero, K.; Cohen, A.; Gburek-Augustat, J.; Synofzik, M.; Brais, B.; Stevens, C.A.; Sanchez-Carpintero, R.; et al. Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J. Med. Genet. 2013, 50, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Wambach, J.A.; Wegner, D.J.; Patni, N.; Kircher, M.; Willing, M.C.; Baldridge, D.; Xing, C.; Agarwal, A.K.; Vergano, S.A.S.; Patel, C.; et al. Bi-allelic POLR3A Loss-of-Function Variants Cause Autosomal-Recessive Wiedemann-Rautenstrauch Syndrome. Am. J. Hum. Genet. 2018, 103, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.orpha.net/ (accessed on 26 November 2023).
- Jay, A.M.; Conway, R.L.; Thiffault, I.; Saunders, C.; Farrow, E.; Adams, J.; Toriello, H.V. Neonatal progeriod syndrome associated with biallelic truncating variants in POLR3A. Am. J. Med. Genet. A 2016, 170, 3343–3346. [Google Scholar] [CrossRef] [PubMed]
- Paolacci, S.; Li, Y.; Agolini, E.; Bellacchio, E.; Arboleda-Bustos, C.E.; Carrero, D.; Bertola, D.; Al-Gazali, L.; Alders, M.; Altmüller, J.; et al. Specific combinations of biallelic POLR3A variants cause Wiedemann-Rautenstrauch syndrome. J. Med. Genet. 2018, 55, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Vanderver, A.; van Spaendonk, R.M.; Schiffmann, R.; Brais, B.; Bugiani, M.; Sistermans, E.; Catsman-Berrevoets, C.; Kros, J.M.; Pinto, P.S.; et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014, 83, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Harting, I.; Al-Saady, M.; Krägeloh-Mann, I.; Bley, A.; Hempel, M.; Bierhals, T.; Karch, S.; Moog, U.; Bernard, G.; Huntsman, R.; et al. POLR3A variants with striatal involvement and extrapyramidal movement disorder. Neurogenetics 2020, 21, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Murtazina, A.F.; Markova, T.V.; Orlova, A.A.; Ryzhkova, O.P.; Shchagina, O.A.; Dadali, E.L. POLR3A-related hypomyelinating leukodystrophy: Case report and literature review. Nervn.-Myshechnye Bolezn. = Neuromuscul. Dis. 2021, 11, 48–54. [Google Scholar] [CrossRef]
- Bernard, G.; Chouery, E.; Putorti, M.L.; Tétreault, M.; Takanohashi, A.; Carosso, G.; Clément, I.; Boespflug-Tanguy, O.; Rodriguez, D.; Delague, V.; et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011, 89, 415–423, Erratum in Am. J. Hum. Genet. 2012, 91, 972. [Google Scholar] [CrossRef] [PubMed]
- Minnerop, M.; Kurzwelly, D.; Rattay, T.W.; Timmann, D.; Hengel, H.; Synofzik, M.; Stendel, C.; Horvath, R.; Schüle, R.; Ramirez, A. Reply: POLR3A variants in hereditary spastic paraplegia and ataxia. Brain 2018, 141, e2. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, I.; Gallo, A.; Ricca, I.; Fini, N.; Silvestri, G.; Gurrieri, F.; Cirillo, M.; Cerase, A.; Natale, G.; Matrone, F.; et al. POLR3A variants in hereditary spastic paraparesis and ataxia: Clinical, genetic, and neuroradiological findings in a cohort of Italian patients. Neurol. Sci. 2022, 43, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- La Piana, R.; Cayami, F.K.; Tran, L.T.; Guerrero, K.; van Spaendonk, R.; Õunap, K.; Pajusalu, S.; Haack, T.; Wassmer, E.; Timmann, D.; et al. Diffuse hypomyelination is not obligate for POLR3-related disorders. Neurology 2016, 86, 1622–1626. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://gnomad.broadinstitute.org/variant/10-78009515-C-T?dataset=gnomad_r4 (accessed on 15 January 2024).
- Minnerop, M.; Kurzwelly, D.; Wagner, H.; Soehn, A.S.; Reichbauer, J.; Tao, F.; Rattay, T.W.; Peitz, M.; Rehbach, K.; Giorgetti, A.; et al. Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain 2017, 140, 1561–1578, Erratum in Brain 2017, 141, e21. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Characterization |
|---|---|
| Current Age | 7.5 years |
| Sex | Female |
| Pregnancy and delivery | Profound IUGR from the 24th week. Delivery at 37 weeks of gestation via cesarean section |
| Birth parameters | A weight deficit (1840 g), body length of 46 cm, and an evident lack of subcutaneous fat |
| Craniofacial features | Progeroid facial features, a hydrocephalic shape of the cranium with a prominent venous network across all regions, micrognathia of the lower jaw, and a defect in the posterior sections of the foramen magnum |
| Dental abnormalities | A neonate tooth (one upper incisor) spontaneously falling out on the 2nd day of life. |
| Postnatal growth | Severe growth retardation (height: 103 cm, SDS: 3.41; growth rate: 3.64 cm/year, SDS: 2.47) and profound body weight deficiency (weight: 10.35 kg, BMI: 9.71 kg/m2, SDS: 6.20). |
| Fat tissue distribution | A general lipodystrophy with distinctive local accumulations of fat (neck, external genitalia, coccygeal region, and feet) |
| Skin findings | A transverse palm crease on the right hand |
| Bone and joint findings | Hip joints and interphalangeal joints contractures, scoliosis, hands and knees joint flexion, and osteoporosis |
| Neurologic and developmental abnormalities | A congenital osteo-neural malformation of the craniocervical junction, an Arnold–Chiari malformation type 1, spina bifida C1, and dysarthria. No intellectual disability |
| Vision and hearing | A retinal angiopathy, an astigmatism, an entropion, and an opacity of the left eye cornea |
| Additional findings | Reactive pancreas changes, ileal lymphofollicular hyperplasia, and steatohepatosis |
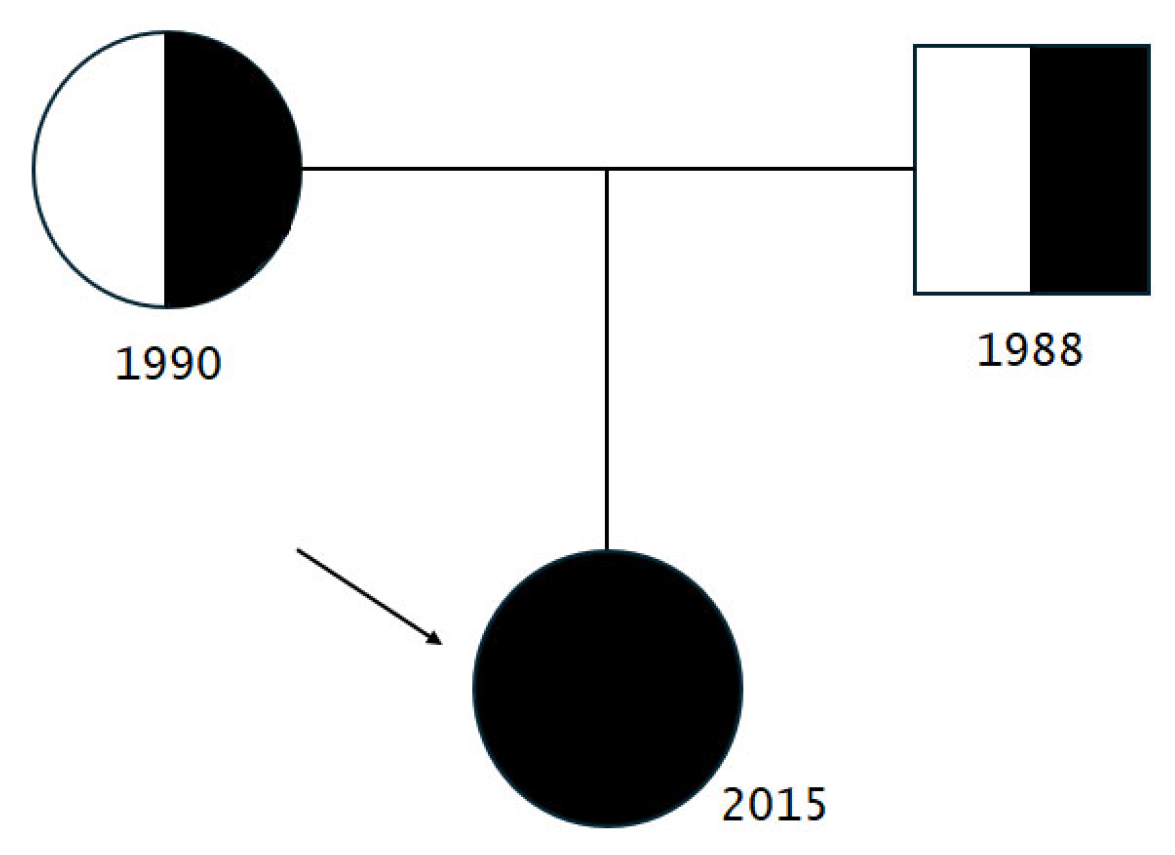
| Family history | Non-consanguineous healthy Russian parents and no siblings |
| POLR3A variants | c.[3677T>C];[3337-11T>C;1909+22G>A] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovalskaia, V.A.; Kungurtseva, A.L.; Bostanova, F.M.; Vasiliev, P.A.; Tabakov, V.Y.; Orlova, M.D.; Povolotskaya, I.S.; Novoselova, O.G.; Bikanov, R.A.; Akhyamova, M.A.; et al. The Genetic Basis of the First Patient with Wiedemann–Rautenstrauch Syndrome in the Russian Federation. Genes 2024, 15, 180. https://doi.org/10.3390/genes15020180
Kovalskaia VA, Kungurtseva AL, Bostanova FM, Vasiliev PA, Tabakov VY, Orlova MD, Povolotskaya IS, Novoselova OG, Bikanov RA, Akhyamova MA, et al. The Genetic Basis of the First Patient with Wiedemann–Rautenstrauch Syndrome in the Russian Federation. Genes. 2024; 15(2):180. https://doi.org/10.3390/genes15020180
Chicago/Turabian StyleKovalskaia, Valeriia A., Anastasiia L. Kungurtseva, Fatima M. Bostanova, Peter A. Vasiliev, Vyacheslav Y. Tabakov, Mariia D. Orlova, Inna S. Povolotskaya, Olga G. Novoselova, Roman A. Bikanov, Mariia A. Akhyamova, and et al. 2024. "The Genetic Basis of the First Patient with Wiedemann–Rautenstrauch Syndrome in the Russian Federation" Genes 15, no. 2: 180. https://doi.org/10.3390/genes15020180
APA StyleKovalskaia, V. A., Kungurtseva, A. L., Bostanova, F. M., Vasiliev, P. A., Tabakov, V. Y., Orlova, M. D., Povolotskaya, I. S., Novoselova, O. G., Bikanov, R. A., Akhyamova, M. A., Tikhonovich, Y. V., Popovich, A. V., Vitebskaya, A. V., Dadali, E. L., & Ryzhkova, O. P. (2024). The Genetic Basis of the First Patient with Wiedemann–Rautenstrauch Syndrome in the Russian Federation. Genes, 15(2), 180. https://doi.org/10.3390/genes15020180