
NF-?B: Governing Macrophages in Cancer

 , , , ,
, , , ,
Abstract
:1. Introduction
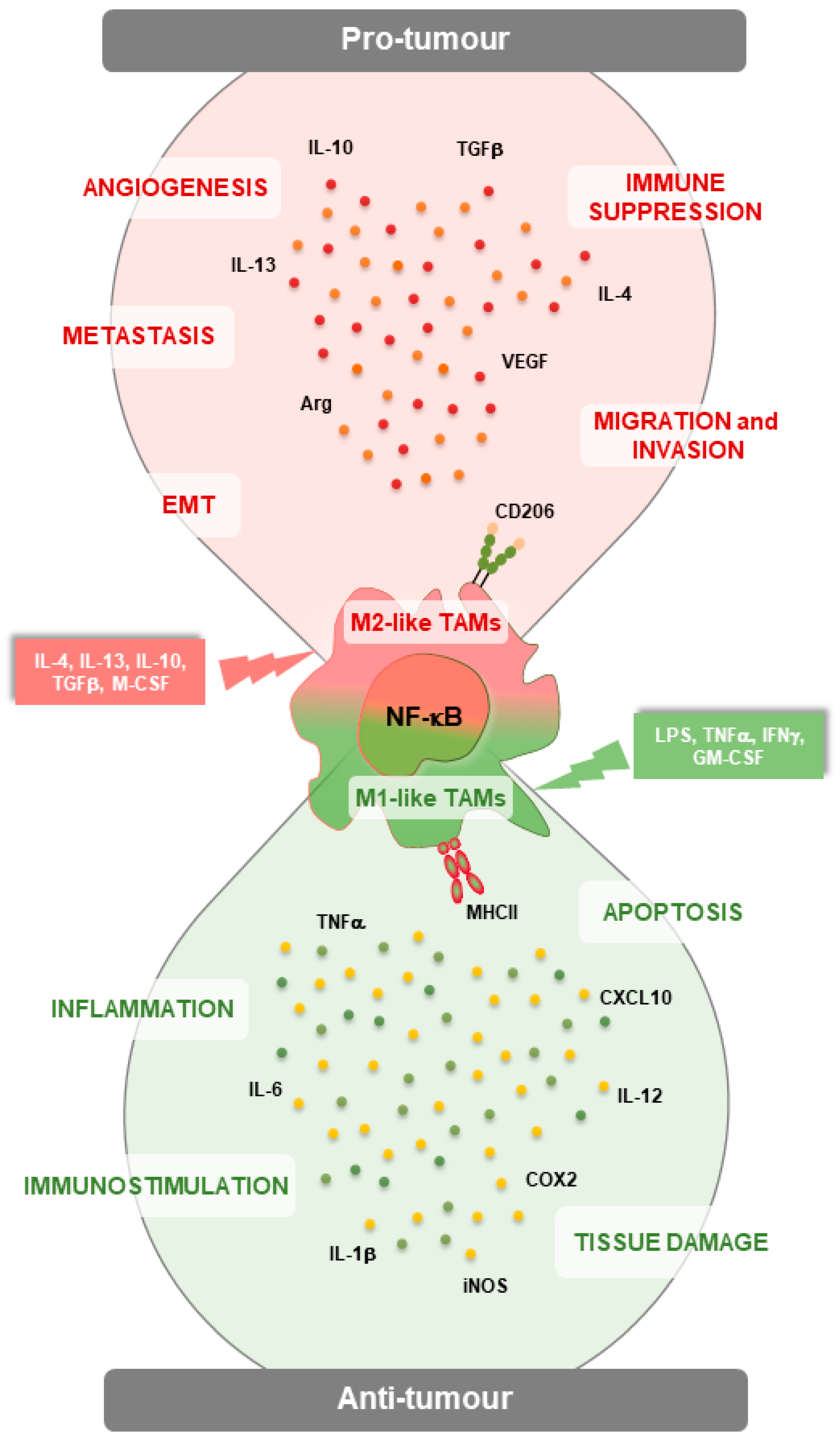
2. The NF-κB Pathway in TAMs

{kind=link}
| Tumor/Cell Type | Stimuli | NF-κB Pathway Component | TAMs Phenotype | Effect | Ref. |
|---|---|---|---|---|---|
| Breast carcinoma | SP1 | p65/p50 | M2 | Pro-tumor | [54,55,56] |
| Melanoma | Mechanical stretch | p65 | M1 | Anti-tumor | [57,58] |
| Lung cancer | TRAIL | p65 | M1 | Anti-tumor | [59] |
| Melanoma, Bladder cancer | HSPs/TLR4 | NF-κB | M2 | Pro-tumor | [60] |
| Breast cancer | M-CSF | p50 | M2 | Pro-tumor | [62] |
| Pancreatic cancer | CUX1 | p65 (RelA) | M2 | Pro-tumor | [66] |
| Hepatoma | TLR2 | p65 | M2 | Pro-tumor (Autophagy) | [67] |
| Pancreatic cancer | IL-33 | IκBα | M2 | Pro-tumor (Dissemination/ Metastasis) | [68,69] |
| Hepatocellular carcinoma | Hypoxia | IKKβ | M2 | Pro-tumor (Angiogenesis, EMT) | [71] |
| hTHP1/mBMDM (Human monocytic cell line/ murine bone marrow-derived macrophage) | Cycling hypoxia | p65 | M1 | Anti-tumor (M0-M1 transition) | [75] |
| Ovarian cancer | Chemotherapeutic agents | IKK | M2 | Pro-tumor | [77] |
| THP1 (monocytic cell line) | Low radiation doses | p50/p50; p65 | M2 | Pro-tumor (Reduced M1 cytokines) | [79] |
| THP1 (monocytic cell line) | Moderate radiation doses | p65/p50 | M1 | Anti-tumor | [80] |
| Mammary Carcinoma, Pancreatic adenocarcinoma | High radiation doses | p50 | M2 | Pro-tumor | [81] |
3. NF-κB Signaling in TAMs: The Lesson from Different Human Cancer Types
3.1. Hepatocellular Carcinoma
3.2. Breast Cancer
3.3. Colon Cancer
3.4. Glioblastoma
3.5. Gynecologic Cancer
3.6. Other Solid Tumors
4. Targeting NF-κB Pathway in TAMs
4.1. Re-Educating TAMs via NF-κB Modulation
4.2. TAMs Depletion and Termination of Recruitment via NF-κB Modulation
4.3. Targeting TAMs in Clinical Setting
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef]
- Verschoor, C.P.; Puchta, A.; Bowdish, D.M.E. The Macrophage. Methods Mol. Biol. 2012, 844, 139–156. [Google Scholar] [CrossRef]
- Taylor, P.R.; Gordon, S. Monocyte Heterogeneity and Innate Immunity. Immunity 2003, 19, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M.; Fusenig, N.E. Friends or Foes—Bipolar Effects of the Tumour Stroma in Cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, A.R.; Greenland, E.L.; Pixley, F.J. Promotion of Tumor Invasion by Tumor-Associated Macrophages: The Role of CSF-1-Activated Phosphatidylinositol 3 Kinase and Src Family Kinase Motility Signaling. Cancers 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.C.; Scott, C.L.; Uronen-Hansson, H.; Gudjonsson, S.; Jansson, O.; Grip, O.; Guilliams, M.; Malissen, B.; Agace, W.W.; Mowat, A.M. Resident and Pro-Inflammatory Macrophages in the Colon Represent Alternative Context-Dependent Fates of the Same Ly6Chi Monocyte Precursors. Mucosal Immunol. 2013, 6, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chintala, N.K.; Vadrevu, S.K.; Patel, J.; Karbowniczek, M.; Markiewski, M.M. Pulmonary Alveolar Macrophages Contribute to the Premetastatic Niche by Suppressing Antitumor T Cell Responses in the Lungs. J. Immunol. 2015, 194, 5529–5538. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1-M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Hagemann, T.; Biswas, S.K.; Lawrence, T.; Sica, A.; Lewis, C.E. Regulation of Macrophage Function in Tumors: The Multifaceted Role of NF-ΚB. Blood 2009, 113, 3139–3146. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Lewis, C.E. NF-ΚB as a Central Regulator of Macrophage Function in Tumors. J. Leukoc. Biol. 2010, 88, 877–884. [Google Scholar] [CrossRef]
- Mulder, R.; Banete, A.; Basta, S. Spleen-Derived Macrophages Are Readily Polarized into Classically Activated (M1) or Alternatively Activated (M2) States. Immunobiology 2014, 219, 737–745. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-X.; Zhang, S.-X.; Wu, H.-J.; Rong, X.-L.; Guo, J. M2b Macrophage Polarization and Its Roles in Diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient Clearance of Early Apoptotic Cells by Human Macrophages Requires M2c Polarization and MerTK Induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef]
- Graff, J.W.; Dickson, A.M.; Clay, G.; McCaffrey, A.P.; Wilson, M.E. Identifying Functional MicroRNAs in Macrophages with Polarized Phenotypes. J. Biol. Chem. 2012, 287, 21816–21825. [Google Scholar] [CrossRef]
- Kaikkonen, M.U.; Lam, M.T.Y.; Glass, C.K. Non-Coding RNAs as Regulators of Gene Expression and Epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, Q.; Wang, X.; Yao, X.; Zhang, B.; Wu, J.; Sun, C. Exosomal NcRNAs Facilitate Interactive “dialogue” between Tumor Cells and Tumor-Associated Macrophages. Cancer Lett. 2023, 552, 215975. [Google Scholar] [CrossRef]
- Ludwig, N.; Rubenich, D.S.; Zaręba, Ł.; Siewiera, J.; Pieper, J.; Braganhol, E.; Reichert, T.E.; Szczepański, M.J. Potential Roles of Tumor Cell- and Stroma Cell-Derived Small Extracellular Vesicles in Promoting a Pro-Angiogenic Tumor Microenvironment. Cancers 2020, 12, 3599. [Google Scholar] [CrossRef] [PubMed]
- Bowdridge, S.; Gause, W.C. Regulation of Alternative Macrophage Activation by Chromatin Remodeling. Nat. Immunol. 2010, 11, 879–881. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-W.; Karin, M. A Cytokine-Mediated Link between Innate Immunity, Inflammation, and Cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the Chemokine Network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 Promotes Tumour Incidence and Growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Vogel, D.Y.S.; Vereyken, E.J.F.; Glim, J.E.; Heijnen, P.D.A.M.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in Inflammatory Multiple Sclerosis Lesions Have an Intermediate Activation Status. J. Neuroinflamm. 2013, 10, 35. [Google Scholar] [CrossRef]
- Pettersen, J.S.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Pierson, K.C.; Pitts-Kiefer, A.; Fan, L.; Belkin, D.A.; Wang, C.Q.F.; Bhuvanendran, S.; Johnson-Huang, L.M.; et al. Tumor-Associated Macrophages in the Cutaneous SCC Microenvironment Are Heterogeneously Activated. J. Investig. Dermatol. 2011, 131, 1322–1330. [Google Scholar] [CrossRef]
- Kerneur, C.; Cano, C.E.; Olive, D. Major Pathways Involved in Macrophage Polarization in Cancer. Front. Immunol. 2022, 13, 1026954. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yang, L.; Cai, J.; Li, H.; Xing, Z.; Hou, Y. Phosphoinositide 3-Kinase/Akt and Its Related Signaling Pathways in the Regulation of Tumor-Associated Macrophages Polarization. Mol. Cell. Biochem. 2022, 477, 2469–2480. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Verzella, D.; Flati, I.; Arboretto, P.; Cornice, J.; Franzoso, G. NF-ΚB: Blending Metabolism, Immunity, and Inflammation. Trends Immunol. 2022, 43, 757–775. [Google Scholar] [CrossRef] [PubMed]
- Moretti, M.; Bennett, J.; Tornatore, L.; Thotakura, A.K.; Franzoso, G. Cancer: NF-ΚB Regulates Energy Metabolism. Int. J. Biochem. Cell Biol. 2012, 44, 2238–2243. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-KappaB: A Key Role in Inflammatory Diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Henkel, T. Function and Activation of NF-Kappa B in the Immune System. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Papa, S.; Franzoso, G.; Sen, R. NF-kappaB-Dependent Regulation of the Timing Of. J. Immunol. 2006, 174, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-ΚB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NF-ΚB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines 2017, 5, 50. [Google Scholar] [CrossRef]
- Verzella, D.; Cornice, J.; Arboretto, P.; Vecchiotti, D.; Di Vito Nolfi, M.; Capece, D.; Zazzeroni, F.; Franzoso, G. The NF-ΚB Pharmacopeia: Novel Strategies to Subdue an Intractable Target. Biomedicines 2022, 10, 2233. [Google Scholar] [CrossRef]
- Bennett, J.; Capece, D.; Begalli, F.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. NF-ΚB in the Crosshairs: Rethinking an Old Riddle. Int. J. Biochem. Cell Biol. 2018, 95, 108–112. [Google Scholar] [CrossRef]
- Hinz, M.; Scheidereit, C. The IκB Kinase Complex in NF-ΚB Regulation and Beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-KappaB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Mitxitorena, I.; Carmody, R.J. The Ubiquitination of NF-ΚB Subunits in the Control of Transcription. Cells 2016, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-ΚB, the First Quarter-Century: Remarkable Progress and Outstanding Questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, S.-C. NF-ΚB in Inflammation and Renal Diseases. Cell Biosci. 2015, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Gilmore, T.D. Good Cop, Bad Cop: The Different Faces of NF-KappaB. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. Non-Canonical NF-ΚB Signaling Pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Kriete, A.; Mayo, K.L. Atypical Pathways of NF-KappaB Activation and Aging. Exp. Gerontol. 2009, 44, 250–255. [Google Scholar] [CrossRef]
- O’Neill, L.A.J. How Toll-like Receptors Signal: What We Know and What We Don’t Know. Curr. Opin. Immunol. 2006, 18, 3–9. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like Receptor Downstream Signaling. Arthritis Res. Ther. 2005, 7, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Tsujimura, H.; Tamura, T. Toll-like Receptor Signaling and Regulation of Cytokine Gene Expression in the Immune System. Biotechniques 2002, 33, S66–S75. [Google Scholar] [CrossRef]
- Weigert, A.; Tzieply, N.; von Knethen, A.; Johann, A.M.; Schmidt, H.; Geisslinger, G.; Brüne, B. Tumor Cell Apoptosis Polarizes Macrophages Role of Sphingosine-1-Phosphate. Mol. Biol. Cell 2007, 18, 3810–3819. [Google Scholar] [CrossRef]
- Rauh, M.J.; Ho, V.; Pereira, C.; Sham, A.; Sly, L.M.; Lam, V.; Huxham, L.; Minchinton, A.I.; Mui, A.; Krystal, G. SHIP Represses the Generation of Alternatively Activated Macrophages. Immunity 2005, 23, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Gangi, L.; Paul, S.; Schioppa, T.; Saccani, A.; Sironi, M.; Bottazzi, B.; Doni, A.; Vincenzo, B.; Pasqualini, F.; et al. A Distinct and Unique Transcriptional Program Expressed by Tumor-Associated Macrophages (Defective NF-KappaB and Enhanced IRF-3/STAT1 Activation). Blood 2006, 107, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, S.; Liu, S.; Li, Z.; Liu, X.; Wu, J. Baicalein Potentiated M1 Macrophage Polarization in Cancer Through Targeting PI3Kγ/ NF-ΚB Signaling. Front. Pharmacol. 2021, 12, 743837. [Google Scholar] [CrossRef]
- Shan, S.; Fang, B.; Zhang, Y.; Wang, C.; Zhou, J.; Niu, C.; Gao, Y.; Zhao, D.; He, J.; Wang, J.; et al. Mechanical Stretch Promotes Tumoricidal M1 Polarization via the FAK/NF-ΚB Signaling Pathway. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 13254–13266. [Google Scholar] [CrossRef]
- Gao, J.; Wang, D.; Liu, D.; Liu, M.; Ge, Y.; Jiang, M.; Liu, Y.; Zheng, D. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Induces the Expression of Proinflammatory Cytokines in Macrophages and Re-Educates Tumor-Associated Macrophages to an Antitumor Phenotype. Mol. Biol. Cell 2015, 26, 3178–3189. [Google Scholar] [CrossRef]
- Lee, C.-H.; Wu, C.-L.; Shiau, A.-L. Toll-like Receptor 4 Signaling Promotes Tumor Growth. J. Immunother. 2010, 33, 73–82. [Google Scholar] [CrossRef]
- He, R.; He, Y.; Du, R.; Liu, C.; Chen, Z.; Zeng, A.; Song, L. Revisiting of TAMs in Tumor Immune Microenvironment: Insight from NF-ΚB Signaling Pathway. Biomed. Pharmacother. 2023, 165, 115090. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Qin, J.; Lan, L.; Li, N.; Wang, C.; He, P.; Liu, F.; Ni, H.; Wang, Y. M-CSF Cooperating with NFκB Induces Macrophage Transformation from M1 to M2 by Upregulating c-Jun. Cancer Biol. Ther. 2014, 15, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Mancino, A.; Lawrence, T. Nuclear Factor-KappaB and Tumor-Associated Macrophages. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 784–789. [Google Scholar] [CrossRef]
- Arkan, M.C.; Greten, F.R. IKK- and NF-ΚB-Mediated Functions in Carcinogenesis. Curr. Top. Microbiol. Immunol. 2011, 349, 159–169. [Google Scholar] [CrossRef]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. P50 Nuclear Factor-ΚB Overexpression in Tumor-Associated Macrophages Inhibits M1 Inflammatory Responses and Antitumor Resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [PubMed]
- Kühnemuth, B.; Michl, P. The Role of CUX1 in Antagonizing NF-ΚB Signaling in TAMs. Oncoimmunology 2014, 3, e28270. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.P.; Su, Y.C.; Hu, C.W.; Lei, H.Y. TLR2-Dependent Selective Autophagy Regulates NF-ΚB Lysosomal Degradation in Hepatoma-Derived M2 Macrophage Differentiation. Cell Death Differ. 2013, 20, 515–523. [Google Scholar] [CrossRef]
- Andersson, P.; Yang, Y.; Hosaka, K.; Zhang, Y.; Fischer, C.; Braun, H.; Liu, S.; Yu, G.; Liu, S.; Beyaert, R.; et al. Molecular Mechanisms of IL-33-Mediated Stromal Interactions in Cancer Metastasis. JCI Insight 2018, 3, e122375. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-Educated Macrophages Promote Tumour Progression and Metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Span, P.N.; Bussink, J. Biology of Hypoxia. Semin. Nucl. Med. 2015, 45, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, L.; Li, X.-F. Hypoxia and the Tumor Microenvironment. Technol. Cancer Res. Treat. 2021, 20, 15330338211036304. [Google Scholar] [CrossRef]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The Inflammatory Microenvironment in Hepatocellular Carcinoma: A Pivotal Role for Tumor-Associated Macrophages. Biomed. Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-KappaB Links Innate Immunity to the Hypoxic Response through Transcriptional Regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A Key Player in Antitumor Immune Response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [PubMed]
- Delprat, V.; Tellier, C.; Demazy, C.; Raes, M.; Feron, O.; Michiels, C. Cycling Hypoxia Promotes a Pro-Inflammatory Phenotype in Macrophages via JNK/P65 Signaling Pathway. Sci. Rep. 2020, 10, 882. [Google Scholar] [CrossRef]
- Medvedeva, G.F.; Kuzmina, D.O.; Nuzhina, J.; Shtil, A.A.; Dukhinova, M.S. How Macrophages Become Transcriptionally Dysregulated: A Hidden Impact of Antitumor Therapy. Int. J. Mol. Sci. 2021, 22, 2662. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, E.M.; Heusinkveld, M.; Tummers, B.; Vogelpoel, L.T.C.; Goedemans, R.; Jha, V.; Nortier, J.W.R.; Welters, M.J.P.; Kroep, J.R.; van der Burg, S.H. Chemotherapy Alters Monocyte Differentiation to Favor Generation of Cancer-Supporting M2 Macrophages in the Tumor Microenvironment. Cancer Res. 2013, 73, 2480–2492. [Google Scholar] [CrossRef] [PubMed]
- Beach, C.; MacLean, D.; Majorova, D.; Arnold, J.N.; Olcina, M.M. The Effects of Radiation Therapy on the Macrophage Response in Cancer. Front. Oncol. 2022, 12, 1020606. [Google Scholar] [CrossRef] [PubMed]
- Lödermann, B.; Wunderlich, R.; Frey, S.; Schorn, C.; Stangl, S.; Rödel, F.; Keilholz, L.; Fietkau, R.; Gaipl, U.S.; Frey, B. Low Dose Ionising Radiation Leads to a NF-ΚB Dependent Decreased Secretion of Active IL-1β by Activated Macrophages with a Discontinuous Dose-Dependency. Int. J. Radiat. Biol. 2012, 88, 727–734. [Google Scholar] [CrossRef]
- Genard, G.; Wera, A.C.; Huart, C.; Le Calve, B.; Penninckx, S.; Fattaccioli, A.; Tabarrant, T.; Demazy, C.; Ninane, N.; Heuskin, A.C.; et al. Proton Irradiation Orchestrates Macrophage Reprogramming through NFκB Signaling. Cell Death Dis. 2018, 9, 728. [Google Scholar] [CrossRef]
- Crittenden, M.R.; Cottam, B.; Savage, T.; Nguyen, C.; Newell, P.; Gough, M.J. Expression of NF-ΚB P50 in Tumor Stroma Limits the Control of Tumors by Radiation Therapy. PLoS ONE 2012, 7, e39295. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bruix, J.; Reig, M.; Sherman, M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 835–853. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-M.; Jing, Y.-Y.; Yu, G.-F.; Kou, X.-R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.-D.; Yang, Y.; Lu, Z.-H.; et al. Tumor-Associated Macrophages Promote Cancer Stem Cell-like Properties via Transforming Growth Factor-Beta1-Induced Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef]
- Budhu, A.; Forgues, M.; Ye, Q.-H.; Jia, H.-L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.-X.; Tang, Z.-Y.; et al. Prediction of Venous Metastases, Recurrence, and Prognosis in Hepatocellular Carcinoma Based on a Unique Immune Response Signature of the Liver Microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.-L.; Leffert, H.; Karin, M. IKKbeta Couples Hepatocyte Death to Cytokine-Driven Compensatory Proliferation That Promotes Chemical Hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-KappaB Functions as a Tumour Promoter in Inflammation-Associated Cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in Liver Parenchymal Cells Causes Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.-Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte Necrosis Induced by Oxidative Stress and IL-1 α Release Mediate Carcinogen-Induced Compensatory Proliferation and Liver Tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef]
- Wei, R.; Zhu, W.W.; Yu, G.Y.; Wang, X.; Gao, C.; Zhou, X.; Lin, Z.F.; Shao, W.Q.; Wang, S.H.; Lu, M.; et al. S100 Calcium-Binding Protein A9 from Tumor-Associated Macrophage Enhances Cancer Stem Cell-like Properties of Hepatocellular Carcinoma. Int. J. Cancer 2021, 148, 1233–1244. [Google Scholar] [CrossRef]
- Fantuzzi, L.; Spadaro, F.; Purificato, C.; Cecchetti, S.; Podo, F.; Belardelli, F.; Gessani, S.; Ramoni, C. Phosphatidylcholine-Specific Phospholipase C Activation Is Required for CCR5-Dependent, NF-KB-Driven CCL2 Secretion Elicited in Response to HIV-1 Gp120 in Human Primary Macrophages. Blood 2008, 111, 3355–3363. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zheng, P.; Wang, W.; Yi, M.; Chen, P.; Cai, J.; Li, J.; Peng, Q.; Ban, Y.; Zhou, Y.; et al. Abberent Expression of NOR1 Protein in Tumor Associated Macrophages Contributes to the Development of DEN-Induced Hepatocellular Carcinoma. J. Cell. Physiol. 2018, 233, 5002–5013. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Zazzeroni, F.; Bubici, C.; Jayawardena, S.; Alvarez, K.; Matsuda, S.; Nguyen, D.U.; Pham, C.G.; Nelsbach, A.H.; Melis, T.; et al. Gadd45 β Mediates the NF-Kappa B Suppression of JNK Signalling by Targeting MKK7/JNKK2. Nat. Cell Biol. 2004, 6, 146–153. [Google Scholar] [CrossRef] [PubMed]
- De Smaele, E.; Zazzeroni, F.; Papa, S.; Nguyen, D.U.; Jin, R.; Jones, J.; Cong, R.; Franzoso, G. Induction of Gadd45beta by NF-KappaB Downregulates pro-Apoptotic JNK Signalling. Nature 2001, 414, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Zazzeroni, F.; Fu, Y.X.; Bubici, C.; Alvarez, K.; Dean, K.; Christiansen, P.A.; Anders, R.A.; Franzoso, G. Gadd45β Promotes Hepatocyte Survival during Liver Regeneration in Mice by Modulating JNK Signaling. J. Clin. Investig. 2008, 118, 1911–1923. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Monti, S.M.; Vitale, R.M.; Bubici, C.; Jayawardena, S.; Alvarez, K.; De Smaele, E.; Dathan, N.; Pedone, C.; Ruvo, M.; et al. Insights into the Structural Basis of the GADD45β-Mediated Inactivation of the JNK Kinase, MKK7/JNKK2. J. Biol. Chem. 2007, 282, 19029–19041. [Google Scholar] [CrossRef]
- Tornatore, L.; Marasco, D.; Dathan, N.; Vitale, R.M.; Benedetti, E.; Papa, S.; Franzoso, G.; Ruvo, M.; Monti, S.M. Gadd45β Forms a Homodimeric Complex That Binds Tightly to MKK7. J. Mol. Biol. 2008, 378, 97–111. [Google Scholar] [CrossRef]
- Verzella, D.; Bennett, J.; Fischietti, M.; Thotakura, A.K.; Recordati, C.; Pasqualini, F.; Capece, D.; Vecchiotti, D.; D’Andrea, D.; Di Francesco, B.; et al. GADD45β Loss Ablates Innate Immunosuppression in Cancer. Cancer Res. 2018, 78, 1275–1292. [Google Scholar] [CrossRef]
- Capece, D.; D’Andrea, D.; Verzella, D.; Tornatore, L.; Begalli, F.; Bennett, J.; Zazzeroni, F.; Franzoso, G. Turning an Old GADDget into a Troublemaker. Cell Death Differ. 2018, 25, 642–644. [Google Scholar] [CrossRef]
- Hu, Y.C.; Yi, Z.J.; Zhou, Y.; Li, P.Z.; Liu, Z.J.; Duan, S.G.; Gong, J.P. Overexpression of RIP 140 Suppresses the Malignant Potential of Hepatocellular Carcinoma by Inhibiting NF-KB-Mediated Alternative Polarization of Macrophages. Oncol. Rep. 2017, 37, 2971–2979. [Google Scholar] [CrossRef]
- Sharen, G.; Cheng, H.; Hu, X.; Miao, J.; Zhao, D. M1-like Tumor-Associated Macrophages Enhance Proliferation and Anti-Apoptotic Ability of Liver Cancer Cells via Activating the NF-ΚB Signaling Pathway. Mol. Med. Rep. 2022, 26, 331. [Google Scholar] [CrossRef]
- Wu, S.-P.; Liao, R.-Q.; Tu, H.-Y.; Wang, W.-J.; Dong, Z.-Y.; Huang, S.-M.; Guo, W.-B.; Gou, L.-Y.; Sun, H.-W.; Zhang, Q.; et al. Stromal PD-L1-Positive Regulatory T Cells and PD-1-Positive CD8-Positive T Cells Define the Response of Different Subsets of Non-Small Cell Lung Cancer to PD-1/PD-L1 Blockade Immunotherapy. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, 521–532. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and Tumor Responses with Lambrolizumab (Anti-PD-1) in Melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Redman, B.G.; Kuzel, T.M.; Harrison, M.R.; Vaishampayan, U.N.; Drabkin, H.A.; George, S.; Logan, T.F.; et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Clark, J.I.; Quinn, D.I. Immune Checkpoint Inhibitors in Advanced Renal Cell Carcinoma: Experience to Date and Future Directions. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective Combinatorial Immunotherapy for Castration-Resistant Prostate Cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef]
- Martini, D.J.; Lalani, A.-K.A.; Bossé, D.; Steinharter, J.A.; Harshman, L.C.; Hodi, F.S.; Ott, P.A.; Choueiri, T.K. Response to Single Agent PD-1 Inhibitor after Progression on Previous PD-1/PD-L1 Inhibitors: A Case Series. J. Immunother. Cancer 2017, 5, 66. [Google Scholar] [CrossRef]
- Xu, G.; Feng, D.; Yao, Y.; Li, P.; Sun, H.; Yang, H.; Li, C.; Jiang, R.; Sun, B.; Chen, Y. Listeria-Based Hepatocellular Carcinoma Vaccine Facilitates Anti-PD-1 Therapy by Regulating Macrophage Polarization. Oncogene 2020, 39, 1429–1444. [Google Scholar] [CrossRef]
- Van den Eynden, G.G.; Majeed, A.W.; Illemann, M.; Vermeulen, P.B.; Bird, N.C.; Høyer-Hansen, G.; Eefsen, R.L.; Reynolds, A.R.; Brodt, P. The Multifaceted Role of the Microenvironment in Liver Metastasis: Biology and Clinical Implications. Cancer Res. 2013, 73, 2031–2043. [Google Scholar] [CrossRef]
- Voorneveld, P.W.; Kodach, L.L.; Jacobs, R.J.; Liv, N.; Zonnevylle, A.C.; Hoogenboom, J.P.; Biemond, I.; Verspaget, H.W.; Hommes, D.W.; de Rooij, K.; et al. Loss of SMAD4 Alters BMP Signaling to Promote Colorectal Cancer Cell Metastasis via Activation of Rho and ROCK. Gastroenterology 2014, 147, 196–208.e13. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lai, X.; Zhao, Y.; Zhang, Y.; Li, M.; Li, D.; Kong, J.; Zhang, Y.; Jing, P.; Li, H.; et al. Loss of NDRG2 in Liver Microenvironment Inhibits Cancer Liver Metastasis by Regulating Tumor Associate Macrophages Polarization Article /13/1 /13/21 /13/31 /13/95 /14/5 /14/19 /38/77 /38/109 /64/60 /82/80. Cell Death Dis. 2018, 9, 248. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Y.S.; Yu, L.G.; Zhang, X.K.; Zhao, L.; Gong, F.L.; Yang, X.X.; Guo, X.L. Galectin-3 Expression and Secretion by Tumor-Associated Macrophages in Hypoxia Promotes Breast Cancer Progression. Biochem. Pharmacol. 2020, 178, 114113. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, W.; Zheng, Y.; Wang, S.; Yang, B.; Li, M.; Song, J.; Zhang, F.; Zhang, X.; Wang, Q.; et al. CXCL1 Derived from Tumor-Associated Macrophages Promotes Breast Cancer Metastasis via Activating NF-ΚB/SOX4 Signaling. Cell Death Dis. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.A.; Kar, S.; Foo, S.L.; Gu, T.; Toh, Y.Q.; Ampomah, P.B.; Sachaphibulkij, K.; Yap, G.; Zharkova, O.; Lukman, H.M.; et al. Annexin-A1 Enhances Breast Cancer Growth and Migration by Promoting Alternative Macrophage Polarization in the Tumour Microenvironment. Sci. Rep. 2017, 7, 17925. [Google Scholar] [CrossRef] [PubMed]
- De Paolis, V.; Maiullari, F.; Chirivì, M.; Milan, M.; Cordiglieri, C.; Pagano, F.; La Manna, A.R.; De Falco, E.; Bearzi, C.; Rizzi, R.; et al. Unusual Association of NF-ΚB Components in Tumor-Associated Macrophages (TAMs) Promotes HSPG2-Mediated Immune-Escaping Mechanism in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 7902. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zürrer-Härdi, U.; Bell, G.; et al. Slug and Sox9 Cooperatively Determine the Mammary Stem Cell State. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Fröse, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A Breast Cancer Stem Cell Niche Supported by Juxtacrine Signalling from Monocytes and Macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Franklin, C.K.; Mackay, A.; Reis-Filho, J.S.; Isacke, C.M. Epithelial and Mesenchymal Subpopulations within Normal Basal Breast Cell Lines Exhibit Distinct Stem Cell/Progenitor Properties. Stem Cells 2012, 30, 292–303. [Google Scholar] [CrossRef]
- Di Vito Nolfi, M.; Vecchiotti, D.; Flati, I.; Verzella, D.; Di Padova, M.; Alesse, E.; Capece, D.; Zazzeroni, F. EV-Mediated Chemoresistance in the Tumor Microenvironment: Is NF-ΚB a Player? Front. Oncol. 2022, 12, 933922. [Google Scholar] [CrossRef]
- Li, C.; Li, R.; Hu, X.; Zhou, G.; Jiang, G. Tumor-Promoting Mechanisms of Macrophage-Derived Extracellular Vesicles-Enclosed MicroRNA-660 in Breast Cancer Progression. Breast Cancer Res. Treat. 2022, 192, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tian, T.; Zhang, J. Tumor-Associated Macrophages (Tams) in Colorectal Cancer (Crc): From Mechanism to Therapy and Prognosis. Int. J. Mol. Sci. 2021, 22, 8470. [Google Scholar] [CrossRef] [PubMed]
- Forssell, J.; Oberg, A.; Henriksson, M.L.; Stenling, R.; Jung, A.; Palmqvist, R. High Macrophage Infiltration along the Tumor Front Correlates with Improved Survival in Colon Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 1472–1479. [Google Scholar] [CrossRef]
- Kang, J.-C.; Chen, J.-S.; Lee, C.-H.; Chang, J.-J.; Shieh, Y.-S. Intratumoral Macrophage Counts Correlate with Tumor Progression in Colorectal Cancer. J. Surg. Oncol. 2010, 102, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Edin, S.; Wikberg, M.L.; Dahlin, A.M.; Rutegård, J.; Öberg, Å.; Oldenborg, P.-A.; Palmqvist, R. The Distribution of Macrophages with a M1 or M2 Phenotype in Relation to Prognosis and the Molecular Characteristics of Colorectal Cancer. PLoS ONE 2012, 7, e47045. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Ippolito, A.; Consonni, F.M.; Carraro, L.; Celesti, G.; Correale, C.; Grizzi, F.; Pasqualini, F.; Tartari, S.; Rinaldi, M.; et al. Protumor Steering of Cancer Inflammation by P50 Nf-Kb Enhances Colorectal Cancer Progression. Cancer Immunol. Res. 2018, 6, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Rawluszko, A.A.; Bujnicka, K.E.; Horbacka, K.; Krokowicz, P.; Jagodziński, P.P. Expression and DNA Methylation Levels of Prolyl Hydroxylases PHD1, PHD2, PHD3 and Asparaginyl Hydroxylase FIH in Colorectal Cancer. BMC Cancer 2013, 13, 526. [Google Scholar] [CrossRef]
- Wang, L.; Niu, Z.; Wang, X.; Li, Z.; Liu, Y.; Luo, F.; Yan, X. PHD2 Exerts Anti-Cancer and Anti-Inflammatory Effects in Colon Cancer Xenografts Mice via Attenuating NF-ΚB Activity. Life Sci. 2020, 242, 117167. [Google Scholar] [CrossRef]
- Capece, D.; D’Andrea, D.; Begalli, F.; Goracci, L.; Tornatore, L.; Alexander, J.L.; Di Veroli, A.; Leow, S.-C.; Vaiyapuri, T.S.; Ellis, J.K.; et al. Enhanced Triacylglycerol Catabolism by Carboxylesterase 1 Promotes Aggressive Colorectal Carcinoma. J. Clin. Investig. 2021, 131(11), e137845. [Google Scholar] [CrossRef]
- Savio, L.E.B.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef]
- Shang, S.; Ji, X.; Zhang, L.; Chen, J.; Li, C.; Shi, R.; Xiang, W.; Kang, X.; Zhang, D.; Yang, F.; et al. Macrophage ABHD5 Suppresses NFκB-Dependent Matrix Metalloproteinase Expression and Cancer Metastasis. Cancer Res. 2019, 79, 5513–5526. [Google Scholar] [CrossRef]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See Chi Ee, P.; San Luis, B.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg Metabolism in Tumor-Conditioned Macrophages Promotes Metastasis in Human Pancreatic Ductal Adenocarcinoma. Oncoimmunology 2016, 5, e1191731. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Steelman, L.S.; Long, J.M.; Kempf, R.C.; Abrams, S.L.; Franklin, R.A.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/MTOR Inhibitors: Rationale and Importance to Inhibiting These Pathways in Human Health. Oncotarget 2011, 2, 135–164. [Google Scholar] [CrossRef] [PubMed]
- Morantz, R.A.; Wood, G.W.; Foster, M.; Clark, M.; Gollahon, K. Macrophages in Experimental and Human Brain Tumors. Part 2: Studies of the Macrophage Content of Human Brain Tumors. J. Neurosurg. 1979, 50, 305–311. [Google Scholar] [CrossRef]
- Chen, G.G.; Chu, Y.S.; Chak, E.C.W.; Leung, B.C.S.; Poon, W.S. Induction of Apoptosis in Glioma Cells by Molecules Released from Activated Macrophages. J. Neurooncol. 2002, 57, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Angara, K.; Jain, M.; Borin, T.F.; Rashid, M.H.; Iskander, A.S.M.; Ara, R.; Kolhe, R.; Howard, S.; Venugopal, N.; et al. Canonical NFκB Signaling in Myeloid Cells Is Required for the Glioblastoma Growth. Sci. Rep. 2017, 7, 13754. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Liang, J.; Wang, W.-Q.; Sun, J.-P.; Udho, E.; Zhang, Z.-Y. PRL3 Promotes Cell Invasion and Proliferation by Down-Regulation of Csk Leading to Src Activation. J. Biol. Chem. 2007, 282, 5413–5419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, L.; Lai, W.; Zeng, Y.; Xu, H.; Lan, Q.; Su, P.; Chu, Z. Interaction with Tumor-associated Macrophages Promotes PRL-3-induced Invasion of Colorectal Cancer Cells via MAPK Pathway-induced EMT and NF-κB Signaling-induced Angiogenesis. Oncol. Rep. 2019, 41, 2790–2802. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Q.; Chen, J.; Zhang, N.; Liu, C.; Wang, T.; Yang, F.; Siebert, H.C.; Zheng, X. Ketogenic Diet Elicits Antitumor Properties through Inducing Oxidative Stress, Inhibiting MMP-9 Expression, and Rebalancing M1/ M2 Tumor-Associated Macrophage Phenotype in a Mouse Model of Colon Cancer. J. Agric. Food Chem. 2020, 68, 11182–11196. [Google Scholar] [CrossRef] [PubMed]
- Lehuédé, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res. 2016, 76, 5201–5208. [Google Scholar] [CrossRef] [PubMed]
- Fehri, E.; Ennaifer, E.; Bel Haj Rhouma, R.; Guizani-Tabbane, L.; Guizani, I.; Boubaker, S. The Role of Toll-like Receptor 9 in Gynecologic Cancer. Curr. Res. Transl. Med. 2016, 64, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Colgan, S.P. Regulation of Immunity and Inflammation by Hypoxia in Immunological Niches. Nat. Rev. Immunol. 2017, 17, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-Dependent Regulation of Inflammatory Pathways in Immune Cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef] [PubMed]
- Chittezhath, M.; Dhillon, M.K.; Lim, J.Y.; Laoui, D.; Shalova, I.N.; Teo, Y.L.; Chen, J.; Kamaraj, R.; Raman, L.; Lum, J.; et al. Molecular Profiling Reveals a Tumor-Promoting Phenotype of Monocytes and Macrophages in Human Cancer Progression. Immunity 2014, 41, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Deng, Y.R.; Wang, Z.C.; Wei, W.F.; Zhou, C.F.; Zhang, Y.M.; Yan, R.M.; Liang, L.J.; Zhong, M.; Liang, L.; et al. Correction: Hypoxia-Induced ZEB1 Promotes Cervical Cancer Progression via CCL8-Dependent Tumour-Associated Macrophage Recruitment. Cell Death Dis. 2022, 13, 247. [Google Scholar] [CrossRef] [PubMed]
- Vitale, C.; Bottino, C.; Castriconi, R. Monocyte and Macrophage in Neuroblastoma: Blocking Their Pro-Tumoral Functions and Strengthening Their Crosstalk with Natural Killer Cells. Cells 2023, 12, 885. [Google Scholar] [CrossRef]
- Leblond, M.M.; Zdimerova, H.; Desponds, E.; Verdeil, G. Tumor-Associated Macrophages in Bladder Cancer: Biological Role, Impact on Therapeutic Response and Perspectives for Immunotherapy. Cancers 2021, 13, 4712. [Google Scholar] [CrossRef]
- Sun, J.; Park, C.; Guenthner, N.; Gurley, S.; Zhang, L.; Lubben, B.; Adebayo, O.; Bash, H.; Chen, Y.; Maksimos, M.; et al. Tumor-Associated Macrophages in Multiple Myeloma: Advances in Biology and Therapy. J. Immunother. Cancer 2022, 10, e003975. [Google Scholar] [CrossRef]
- Chen, Y.L. Prognostic Significance of Tumor-Associated Macrophages in Patients with Nasopharyngeal Carcinoma: A Meta-Analysis. Medicine 2020, 99, E21999. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Healey, J.; Ogura, K.; Yoshida, A.; Kondo, H.; Hata, T.; Kure, M.; Tazawa, H.; Nakata, E.; Kunisada, T.; et al. Role of Tumor-Associated Macrophages in Sarcomas. Cancers 2021, 13, 1086. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2021, 8, 607209. [Google Scholar] [CrossRef]
- Lankadasari, M.B.; Mukhopadhyay, P.; Mohammed, S.; Harikumar, K.B. TAMing Pancreatic Cancer: Combat with a Double Edged Sword. Mol. Cancer 2019, 18, 48. [Google Scholar] [CrossRef] [PubMed]
- XU, F.; WEI, Y.; TANG, Z.; LIU, B.; DONG, J. Tumor-Associated Macrophages in Lung Cancer: Friend or Foe? Mol. Med. Rep. 2020, 22, 4107–4115. [Google Scholar]
- Shen, H.; Liu, J.; Chen, S.; Ma, X.; Ying, Y.; Li, J.; Wang, W.; Wang, X.; Xie, L. Prognostic Value of Tumor-Associated Macrophages in Clear Cell Renal Cell Carcinoma: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 657318. [Google Scholar] [CrossRef] [PubMed]
- Rihawi, K.; Ricci, A.D.; Rizzo, A.; Brocchi, S.; Marasco, G.; Pastore, L.V.; Llimpe, F.L.R.; Golfieri, R.; Renzulli, M. Tumor-Associated Macrophages and Inflammatory Microenvironment in Gastric Cancer: Novel Translational Implications. Int. J. Mol. Sci. 2021, 22, 3805. [Google Scholar] [CrossRef]
- Gambardella, V.; Castillo, J.; Tarazona, N.; Gimeno-Valiente, F.; Martínez-Ciarpaglini, C.; Cabeza-Segura, M.; Roselló, S.; Roda, D.; Huerta, M.; Cervantes, A.; et al. The Role of Tumor-Associated Macrophages in Gastric Cancer Development and Their Potential as a Therapeutic Target. Cancer Treat. Rev. 2020, 86, 102015. [Google Scholar] [CrossRef]
- Fujimura, T.; Kambayashi, Y.; Fujisawa, Y.; Hidaka, T.; Aiba, S. Tumor-Associated Macrophages: Therapeutic Targets for Skin Cancer. Front. Oncol. 2018, 8, 3. [Google Scholar] [CrossRef]
- Chiang, E.; Stafford, H.; Buell, J.; Ramesh, U.; Amit, M.; Nagarajan, P.; Migden, M.; Yaniv, D. Review of the Tumor Microenvironment in Basal and Squamous Cell Carcinoma. Cancers 2023, 15, 2453. [Google Scholar] [CrossRef]
- Zhou, Q.; Fang, T.; Wei, S.; Chai, S.; Yang, H.; Tao, M.; Cao, Y. Macrophages in Melanoma: A Double-edged Sword and Targeted Therapy Strategies (Review). Exp. Ther. Med. 2022, 24, 640. [Google Scholar] [CrossRef]
- Xue, Y.; Song, X.; Fan, S.; Deng, R. The Role of Tumor-Associated Macrophages in Oral Squamous Cell Carcinoma. Front. Physiol. 2022, 13, 959747. [Google Scholar] [CrossRef]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef]
- Kumari, N.; Choi, S.H. Tumor-Associated Macrophages in Cancer: Recent Advancements in Cancer Nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41, 68. [Google Scholar] [CrossRef]
- Qiu, B.; Matthay, K.K. Advancing Therapy for Neuroblastoma. Nat. Rev. Clin. Oncol. 2022, 19, 515–533. [Google Scholar] [CrossRef]
- Zhang, Q.; Mao, Z.; Sun, J. NF-ΚB Inhibitor, BAY11-7082, Suppresses M2 Tumor-Associated Macrophage Induced EMT Potential via MiR-30a/NF-ΚB/Snail Signaling in Bladder Cancer Cells. Gene 2019, 710, 91–97. [Google Scholar] [CrossRef]
- Garcia, J.A.; Cowey, C.L.; Godley, P.A. Renal Cell Carcinoma. Curr. Opin. Oncol. 2009, 21, 266–271. [Google Scholar] [CrossRef]
- Mukherjee, S.; Baidoo, J.; Fried, A.; Atwi, D.; Dolai, S.; Boockvar, J.; Symons, M.; Ruggieri, R.; Raja, K.; Banerjee, P. Curcumin Changes the Polarity of Tumor-Associated Microglia and Eliminates Glioblastoma. Int. J. cancer 2016, 139, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Kai, K.; Komohara, Y.; Esumi, S.; Fujiwara, Y.; Yamamoto, T.; Uekawa, K.; Ohta, K.; Takezaki, T.; Kuroda, J.; Shinojima, N.; et al. Macrophage/Microglia-Derived IL-1β Induces Glioblastoma Growth via the STAT3/NF-ΚB Pathway. Hum. Cell 2022, 35, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xu, J.B.; He, Y.L.; Peng, J.J.; Zhang, X.H.; Chen, C.Q.; Li, W.; Cai, S.R. Tumor-Associated Macrophages Promote Angiogenesis and Lymphangiogenesis of Gastric Cancer. J. Surg. Oncol. 2012, 106, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Tjiu, J.W.; Chen, J.S.; Shun, C.T.; Lin, S.J.; Liao, Y.H.; Chu, C.Y.; Tsai, T.F.; Chiu, H.C.; Dai, Y.S.; Inoue, H.; et al. Tumor-Associated Macrophage-Induced Invasion and Angiogenesis of Human Basal Cell Carcinoma Cells by Cyclooxygenase-2 Induction. J. Investig. Dermatol. 2009, 129, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Seijo, A.; García-Martínez, E.; Barrio-Alonso, C.; Pareja-Malagón, M.; Acosta-Ocampo, A.; Fernández-Santos, M.E.; Puig-Kröger, A.; Parra-Blanco, V.; Mercader, E.; Márquez-Rodas, I.; et al. Ccl20/Tnf/Vegfa Cytokine Secretory Phenotype of Tumor-Associated Macrophages Is a Negative Prognostic Factor in Cutaneous Melanoma. Cancers 2021, 13, 3943. [Google Scholar] [CrossRef] [PubMed]
- Paccez, J.D.; Vogelsang, M.; Parker, M.I.; Zerbini, L.F. The Receptor Tyrosine Kinase Axl in Cancer: Biological Functions and Therapeutic Implications. Int. J. Cancer 2014, 134, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, V.A. Axl-Dependent Signalling: A Clinical Update. Clin. Sci. 2012, 122, 361–368. [Google Scholar] [CrossRef]
- Chiu, K.C.; Lee, C.H.; Liu, S.Y.; Chou, Y.T.; Huang, R.Y.; Huang, S.M.; Shieh, Y.S. Polarization of Tumor-Associated Macrophages and Gas6/Axl Signaling in Oral Squamous Cell Carcinoma. Oral Oncol. 2015, 51, 683–689. [Google Scholar] [CrossRef]
- Lee, C.H.; Liu, S.Y.; Chou, K.C.; Yeh, C.T.; Shiah, S.G.; Huang, R.Y.; Cheng, J.C.; Yen, C.Y.; Shieh, Y.S. Tumor-Associated Macrophages Promote Oral Cancer Progression through Activation of the Axl Signaling Pathway. Ann. Surg. Oncol. 2014, 21, 1031–1037. [Google Scholar] [CrossRef]
- Vecchiotti, D.; Verzella, D.; Di Vito Nolfi, M.; D’andrea, D.; Flati, I.; Di Francesco, B.; Cornice, J.; Alesse, E.; Capece, D.; Zazzeroni, F. Elevated NF-ΚB/SHh/GLI1 Signature Denotes a Worse Prognosis and Represent a Novel Potential Therapeutic Target in Advanced Prostate Cancer. Cells 2022, 11, 2118. [Google Scholar] [CrossRef]
- Chen, P.-C.; Cheng, H.-C.; Wang, J.; Wang, S.-W.; Tai, H.-C.; Lin, C.-W.; Tang, C.-H. Prostate Cancer-Derived CCN3 Induces M2 Macrophage Infiltration and Contributes to Angiogenesis in Prostate Cancer Microenvironment. Oncotarget 2014, 5, 1595. [Google Scholar] [CrossRef]
- Beider, K.; Bitner, H.; Leiba, M.; Gutwein, O.; Koren-Michowitz, M.; Ostrovsky, O.; Abraham, M.; Wald, H.; Galun, E.; Peled, A.; et al. Multiple Myeloma Cells Recruit Tumor-Supportive Macrophages through the CXCR4/CXCL12 Axis and Promote Their Polarization toward the M2 Phenotype. Oncotarget 2014, 5, 11283–11296. [Google Scholar] [CrossRef]
- Khalife, J.; Ghose, J.; Martella, M.; Viola, D.; Rocci, A.; Troadec, E.; Terrazas, C.; Satoskar, A.R.; Gunes, E.G.; Dona, A.; et al. MiR-16 Regulates Crosstalk in NF-ΚB Tolerogenic Inflammatory Signaling between Myeloma Cells and Bone Marrow Macrophages. JCI Insight 2019, 4, e129348. [Google Scholar] [CrossRef]
- Song, Y.; Li, X.; Zeng, Z.; Li, Q.; Gong, Z.; Liao, Q.; Li, X.; Chen, P.; Xiang, B.; Zhang, W.; et al. Epstein-Barr Virus Encoded MiR-BART11 Promotes Inflammation-Induced Carcinogenesis by Targeting FOXP1. Oncotarget 2016, 7, 36783. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An Epigenetic Switch Involving NF-KappaB, Lin28, Let-7 MicroRNA, and IL6 Links Inflammation to Cell Transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hao, Z.; Hong, Y.; He, W.; Zhao, W. Reprogramming Tumor Associated Macrophage Phenotype by a Polysaccharide from Ilex Asprella for Sarcoma Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3816. [Google Scholar] [CrossRef] [PubMed]
- Kühnemuth, B.; Mühlberg, L.; Schipper, M.; Griesmann, H.; Neesse, A.; Milosevic, N.; Wissniowski, T.; Buchholz, M.; Gress, T.M.; Michl, P. CUX1 Modulates Polarization of Tumor-Associated Macrophages by Antagonizing NF-ΚB Signaling. Oncogene 2015, 34, 177–187. [Google Scholar] [CrossRef]
- Kemp, S.B.; Carpenter, E.S.; Steele, N.G.; Donahue, K.L.; Nwosu, Z.C.; Pacheco, A.; Velez-Delgado, A.; Menjivar, R.E.; Lima, F.; The, S.; et al. Apolipoprotein E Promotes Immune Suppression in Pancreatic Cancer through NF-ΚB-Mediated Production of CXCL1. Cancer Res. 2021, 81, 4305–4318. [Google Scholar] [CrossRef]
- Zhu, C.; Shen, H.; Zhu, L.; Zhao, F.; Shu, Y. Plasminogen Activator Inhibitor 1 Promotes Immunosuppression in Human Non-Small Cell Lung Cancers by Enhancing TGF-Β1 Expression in Macrophage. Cell. Physiol. Biochem. 2018, 44, 2201–2211. [Google Scholar] [CrossRef]
- Feng, X.; Yu, W.; Cao, L.; Meng, F.; Cong, M. A Novel Chrysin Thiazole Derivative Polarizes Macrophages to an M1 Phenotype via Targeting TLR4. Int. Immunopharmacol. 2020, 88, 106986. [Google Scholar] [CrossRef]
- Yin, L.; Fan, Z.; Liu, P.; Chen, L.; Guan, Z.; Liu, Y.; Luo, Y. Anemoside A3 Activates TLR4-Dependent M1-Phenotype Macrophage Polarization to Represses Breast Tumor Growth and Angiogenesis. Toxicol. Appl. Pharmacol. 2021, 432, 115755. [Google Scholar] [CrossRef]
- Cheng, H.; Sun, L.; Shen, D.; Ren, A.; Ma, F.; Tai, G.; Fan, L.; Zhou, Y. β-1,6 Glucan Converts Tumor-Associated Macrophages into an M1-like Phenotype. Carbohydr. Polym. 2020, 247, 116715. [Google Scholar] [CrossRef]
- Qian, S.; Han, X.; Sha, X.; Tian, F.; Huang, H.; Jiang, P.; Huang, G.; Ma, B.; Zhang, H.; Zhu, Y.; et al. Aqueous Extract of Cimicifuga Dahurica Reprogramming Macrophage Polarization by Activating TLR4-NF-ΚB Signaling Pathway. J. Inflamm. Res. 2022, 15, 1027–1046. [Google Scholar] [CrossRef]
- Liu, C.; He, D.; Zhang, S.; Chen, H.; Zhao, J.; Li, X.; Zeng, X. Homogeneous Polyporus Polysaccharide Inhibit Bladder Cancer by Resetting Tumor-Associated Macrophages Toward M1 Through NF-ΚB/NLRP3 Signaling. Front. Immunol. 2022, 13, 839460. [Google Scholar] [CrossRef]
- Hoover, A.A.; Hufnagel, D.H.; Harris, W.; Bullock, K.; Glass, E.B.; Liu, E.; Barham, W.; Crispens, M.A.; Khabele, D.; Giorgio, T.D.; et al. Increased Canonical NF-KappaB Signaling Specifically in Macrophages Is Sufficient to Limit Tumor Progression in Syngeneic Murine Models of Ovarian Cancer. BMC Cancer 2020, 20, 970. [Google Scholar] [CrossRef]
- Ai, Y.; Liu, S.; Luo, H.; Wu, S.; Wei, H.; Tang, Z.; Li, X.; Zou, C. LncRNA DCST1-AS1 Facilitates Oral Squamous Cell Carcinoma by Promoting M2 Macrophage Polarization through Activating NF- κ B Signaling. J. Immunol. Res. 2021, 2021, 5524231. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, L.; Li, Z.; Zhang, W.; Luo, F.; Chu, Y.; Chen, G. Glycocalyx-Mimicking Nanoparticles Improve Anti-PD-L1 Cancer Immunotherapy through Reversion of Tumor-Associated Macrophages. Biomacromolecules 2018, 19, 2098–2108. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, S.; Jiang, D.; Gao, T.; Fang, Y.; Fu, S.; Guan, L.; Zhang, Z.; Mu, W.; Chu, Q.; et al. Manipulation of TAMs Functions to Facilitate the Immune Therapy Effects of Immune Checkpoint Antibodies. J. Control. Release 2021, 336, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Parayath, N.N.; Parikh, A.; Amiji, M.M. Repolarization of Tumor-Associated Macrophages in a Genetically Engineered Nonsmall Cell Lung Cancer Model by Intraperitoneal Administration of Hyaluronic Acid-Based Nanoparticles Encapsulating MicroRNA-125b. Nano Lett. 2018, 18, 3571–3579. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Lu, L.; Xue, C.; Liu, J.; He, Y.; Zhou, J.; Xia, Z.; Dai, L.; Luo, Z.; Mao, Y.; et al. Polarization of Tumor-Associated Macrophage Phenotype: Via Porous Hollow Iron Nanoparticles for Tumor Immunotherapy in Vivo. Nanoscale 2020, 12, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.; Guo, X.; Zhao, J.; Zhou, S. A Biomimetic Polymer Magnetic Nanocarrier Polarizing Tumor-Associated Macrophages for Potentiating Immunotherapy. Small 2020, 16, e2003543. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, L.; Zhen, M.; Zhen, M.; Wang, H.; Wang, H.; Sun, Z.; Sun, Z.; Jia, W.; Jia, W.; et al. Functional Gadofullerene Nanoparticles Trigger Robust Cancer Immunotherapy Based on Rebuilding an Immunosuppressive Tumor Microenvironment. Nano Lett. 2020, 20, 4487–4496. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zheng, B.; Zhang, S.; Liao, X.; Tong, Q.; Wei, G.; Yu, S.; Chen, G.; Wu, A.; Gao, S.; et al. Copper Sulfide Nanoparticle-Redirected Macrophages for Adoptive Transfer Therapy of Melanoma. Adv. Funct. Mater. 2021, 31, 2008022. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, X.; Huo, M.; Wang, Y.; Li, Y.; Xu, N.; Zhu, H. Cetuximab Enhances the Anti-Tumor Function of Macrophages in an IL-6 Dependent Manner. Life Sci. 2021, 267, 118953. [Google Scholar] [CrossRef]
- Cao, X.; Li, B.; Chen, J.; Dang, J.; Chen, S.; Gunes, E.G.; Xu, B.; Tian, L.; Muend, S.; Raoof, M.; et al. Effect of Cabazitaxel on Macrophages Improves CD47-Targeted Immunotherapy for Triple-Negative Breast Cancer. J. Immunother. Cancer 2021, 9, e002022. [Google Scholar] [CrossRef]
- Jin, J.; Lin, J.; Xu, A.; Lou, J.; Qian, C.; Li, X.; Wang, Y.; Yu, W.; Tao, H. CCL2: An Important Mediator Between Tumor Cells and Host Cells in Tumor Microenvironment. Front. Oncol. 2021, 11, 722916. [Google Scholar] [CrossRef]
- Jin, L.; Guo, Y.; Mao, W.; Wang, J.; Jin, L.; Liu, X.; Shou, Q.; Fu, H. Total Glucosides of Paeony Inhibit Breast Cancer Growth by Inhibiting TAMs Infiltration through NF-ΚB/CCL2 Signaling. Phytomedicine 2022, 104, 154307. [Google Scholar] [CrossRef]
- Ortega, R.A.; Barham, W.; Sharman, K.; Tikhomirov, O.; Giorgio, T.D.; Yull, F.E. Manipulating the NF-ΚB Pathway in Macrophages Using Mannosylated, SiRNA-Delivering Nanoparticles Can Induce Immunostimulatory and Tumor Cytotoxic Functions. Int. J. Nanomed. 2016, 11, 2163–2177. [Google Scholar] [CrossRef]
- Tsagozis, P.; Augsten, M.; Pisa, P. All Trans-Retinoic Acid Abrogates the pro-Tumorigenic Phenotype of Prostate Cancer Tumor-Associated Macrophages. Int. Immunopharmacol. 2014, 23, 8–13. [Google Scholar] [CrossRef]
- Wanderley, C.W.; Colón, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel Reduces Tumor Growth by Reprogramming Tumor-Associated Macrophages to an M1 Profile in a TLR4-Dependent Manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of Tumour-Infiltrating Macrophages via CCL2/CCR2 Signalling as a Therapeutic Strategy against Hepatocellular Carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef]
- Glass, E.B.; Hoover, A.A.; Bullock, K.K.; Madden, M.Z.; Reinfeld, B.I.; Harris, W.; Parker, D.; Hufnagel, D.H.; Crispens, M.A.; Khabele, D.; et al. Stimulating TAM-Mediated Anti-Tumor Immunity with Mannose-Decorated Nanoparticles in Ovarian Cancer. BMC Cancer 2022, 22, 497. [Google Scholar] [CrossRef]
- Liu, M.; Sakamaki, T.; Casimiro, M.C.; Willmarth, N.E.; Quong, A.A.; Ju, X.; Ojeifo, J.; Jiao, X.; Yeow, W.-S.; Katiyar, S.; et al. The Canonical NF-KappaB Pathway Governs Mammary Tumorigenesis in Transgenic Mice and Tumor Stem Cell Expansion. Cancer Res. 2010, 70, 10464–10473. [Google Scholar] [CrossRef]
- Tornatore, L.; Sandomenico, A.; Raimondo, D.; Low, C.; Rocci, A.; Tralau-Stewart, C.; Capece, D.; D’Andrea, D.; Bua, M.; Boyle, E.; et al. Cancer-Selective Targeting of the NF-ΚB Survival Pathway with GADD45β/MKK7 Inhibitors. Cancer Cell 2014, 26, 495–508. [Google Scholar] [CrossRef]
- Tornatore, L.; Capece, D.; D’Andrea, D.; Begalli, F.; Verzella, D.; Bennett, J.; Acton, G.; Campbell, E.A.; Kelly, J.; Tarbit, M.; et al. Clinical Proof of Concept for a Safe and Effective NF-ΚB-Targeting Strategy in Multiple Myeloma. Br. J. Haematol. 2019, 185, 588–592. [Google Scholar] [CrossRef]
- Tornatore, L.; Capece, D.; D’Andrea, D.; Begalli, F.; Verzella, D.; Bennett, J.; Acton, G.; Campbell, E.A.; Kelly, J.; Tarbit, M.; et al. Preclinical Toxicology and Safety Pharmacology of the First-in-Class GADD45β/MKK7 Inhibitor and Clinical Candidate, DTP3. Toxicol. Rep. 2019, 6, 369–379. [Google Scholar] [CrossRef]
- Rega, C.; Russo, R.; Focà, A.; Sandomenico, A.; Iaccarino, E.; Raimondo, D.; Milanetti, E.; Tornatore, L.; Franzoso, G.; Pedone, P.V.; et al. Probing the Interaction Interface of the GADD45β/MKK7 and MKK7/DTP3 Complexes by Chemical Cross-Linking Mass Spectrometry. Int. J. Biol. Macromol. 2018, 114, 114–123. [Google Scholar] [CrossRef]
- Sandomenico, A.; Di Rienzo, L.; Calvanese, L.; Iaccarino, E.; D’auria, G.; Falcigno, L.; Chambery, A.; Russo, R.; Franzoso, G.; Tornatore, L.; et al. Insights into the Interaction Mechanism of Dtp3 with Mkk7 by Using Std-Nmr and Computational Approaches. Biomedicines 2021, 9, 20. [Google Scholar] [CrossRef]
- Pittet, M.J.; Michielin, O.; Migliorini, D. Clinical Relevance of Tumour-Associated Macrophages. Nat. Rev. Clin. Oncol. 2022, 19, 402–421. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as Tools and Targets in Cancer Therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Ribas, A.; Medina, T.; Kirkwood, J.M.; Zakharia, Y.; Gonzalez, R.; Davar, D.; Chmielowski, B.; Campbell, K.M.; Bao, R.; Kelley, H.; et al. Overcoming PD-1 Blockade Resistance with CpG-A Toll-like Receptor 9 Agonist Vidutolimod in Patients with Metastatic Melanoma. Cancer Discov. 2021, 11, 2998–3007. [Google Scholar] [CrossRef]
- Chow, L.Q.M.; Morishima, C.; Eaton, K.D.; Baik, C.S.; Goulart, B.H.; Anderson, L.N.; Manjarrez, K.L.; Dietsch, G.N.; Bryan, J.K.; Hershberg, R.M.; et al. Phase Ib Trial of the Toll-like Receptor 8 Agonist, Motolimod (VTX-2337), Combined with Cetuximab in Patients with Recurrent or Metastatic SCCHN. Clin. Cancer Res. 2017, 23, 2442–2450. [Google Scholar] [CrossRef]
- Cassier, P.A.; Italiano, A.; Gomez-Roca, C.A.; Le Tourneau, C.; Toulmonde, M.; Cannarile, M.A.; Ries, C.; Brillouet, A.; Müller, C.; Jegg, A.M.; et al. CSF1R Inhibition with Emactuzumab in Locally Advanced Diffuse-Type Tenosynovial Giant Cell Tumours of the Soft Tissue: A Dose-Escalation and Dose-Expansion Phase 1 Study. Lancet Oncol. 2015, 16, 949–956. [Google Scholar] [CrossRef]
- Gomez-Roca, C.; Cassier, P.; Zamarin, D.; Machiels, J.P.; Luis Perez Gracia, J.; Stephen Hodi, F.; Taus, A.; Martinez Garcia, M.; Boni, V.; Eder, J.P.; et al. Anti-CSF-1R Emactuzumab in Combination with Anti-PD-L1 Atezolizumab in Advanced Solid Tumor Patients Naïve or Experienced for Immune Checkpoint Blockade. J. Immunother. Cancer 2022, 10, e004076. [Google Scholar] [CrossRef]
- Gomez-Roca, C.A.; Italiano, A.; Le Tourneau, C.; Cassier, P.A.; Toulmonde, M.; D’Angelo, S.P.; Campone, M.; Weber, K.L.; Loirat, D.; Cannarile, M.A.; et al. Phase i Study of Emactuzumab Single Agent or in Combination with Paclitaxel in Patients with Advanced/Metastatic Solid Tumors Reveals Depletion of Immunosuppressive M2-like Macrophages. Ann. Oncol. 2019, 30, 1381–1392. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting Tumour-Associated Macrophages with CCR2 Inhibition in Combination with FOLFIRINOX in Patients with Borderline Resectable and Locally Advanced Pancreatic Cancer: A Single-Centre, Open-Label, Dose-Finding, Non-Randomised, Phase 1b Trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef]
- Upton, R.; Banuelos, A.; Feng, D.; Biswas, T.; Kao, K.; McKenna, K.; Willingham, S.; Ho, P.Y.; Rosental, B.; Tal, M.C.; et al. Combining CD47 Blockade with Trastuzumab Eliminates HER2-Positive Breast Cancer Cells and Overcomes Trastuzumab Tolerance. Proc. Natl. Acad. Sci. USA 2021, 118, e2026849118. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 Agonistic Monoclonal Antibody APX005M (Sotigalimab) and Chemotherapy, with or without Nivolumab, for the Treatment of Metastatic Pancreatic Adenocarcinoma: An Open-Label, Multicentre, Phase 1b Study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Irenaeus, S.M.M.; Nielsen, D.; Ellmark, P.; Yachnin, J.; Deronic, A.; Nilsson, A.; Norlén, P.; Veitonmäki, N.; Wennersten, C.S.; Ullenhag, G.J. First-in-Human Study with Intratumoral Administration of a CD40 Agonistic Antibody, ADC-1013, in Advanced Solid Malignancies. Int. J. Cancer 2019, 145, 1189–1199. [Google Scholar] [CrossRef]
| TAM REPROGRAMMING | |||
|---|---|---|---|
| Molecules/Drug | Target | Cancers | Ref. |
| Chrysin thiazole derivative (ChR-TD) | TLR4/NF-κB | Breast cancer cell line (4T1) | [187] |
| Anemoside A3 (A3) | TLR4/NF-κB | Breast cancer | [188] |
| β-D-(1→6) glucan (AAMP-A70) | TLR2/Akt/NF-κB | Colon cancer cells | [189] |
| Aqueous Extract of Cimicifuga dahurica (CRAE) | TLR4/MyD88/TAK1/NF-κB | MM | [190] |
| Homogeneous Polyporus Polysaccharide (HPP) | TLR2/NF-κB/NLRP3 | Bladder cancer | [191] |
| Baicalein | PI3K/NF-κB | Breast cancer, melanoma | [57] |
| IKK2 | NF-κB | OC | [192] |
| BAY11-7082 | NF-κB/miR30a | Bladder cancer cells | [166] |
| IncRNA DCST1-AS1 | NF-κB | OSCC | [193] |
| Glycocalyx-mimicking nanoparticles (GNPs) | STAT6 and NF-κB | LLC | [194] |
| Mannose modified lipid nanoparticles (M-IMD-LNP) with IMD-0354 | NF-κB | Melanoma cells (B16) | [195] |
| Hyaluronic acid (HA) nanoparticles, loaded with micro-RNA miR-125 | NF-κB | NSCLC | [196] |
| Porous hollow iron oxide nanoparticles (PHNPs) loaded with 3-methyladenine (3-MA) | PI3Kγ/Akt/NF-κB | Breast cancer cell line (MDA-MB-231) | [197] |
| PLGA-ION-R837@M | TLR7/IRF5/NF-κB | Breast cancer cell line (4T1) | [198] |
| Gd@C82 nanoparticles modified with b-alanines (GF-Ala) | NF-κB/IRF5 | Breast cancer cell line (4T1) | [199] |
| Copper sulfide nanoparticles (CuS-NP) | NF-κB | Melanoma | [200] |
| Cetuximab | NF-κB and STAT3 | CRC | [201] |
| Cabazitaxel | TLR/NF-κB | Breast cancer cells | [202] |
| Proton irradiation | NF-κB | THP1 cells | [80] |
| TAM DEPLETION AND TERMINATION OF RECRUITMENT | |||
| CCR2 antagonist or knocking out of host CCR2 | CCL2/CCR2/NF-κB | HCC | [203] |
| Total glucosides of paeony (TGP) | NF-κB/CCL2 | Breast cancer | [204] |
| IκBα si-RNA encapsulated into mannosylated siRNA-delivering NPs | NF-κB | OC, breast cancer | [205] |
| Trans-retinoic acid (ATRA) | NF-κB | PCa | [206] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornice, J.; Verzella, D.; Arboretto, P.; Vecchiotti, D.; Capece, D.; Zazzeroni, F.; Franzoso, G. NF-?B: Governing Macrophages in Cancer. Genes 2024, 15, 197. https://doi.org/10.3390/genes15020197
Cornice J, Verzella D, Arboretto P, Vecchiotti D, Capece D, Zazzeroni F, Franzoso G. NF-?B: Governing Macrophages in Cancer. Genes. 2024; 15(2):197. https://doi.org/10.3390/genes15020197
Chicago/Turabian StyleCornice, Jessica, Daniela Verzella, Paola Arboretto, Davide Vecchiotti, Daria Capece, Francesca Zazzeroni, and Guido Franzoso. 2024. "NF-?B: Governing Macrophages in Cancer" Genes 15, no. 2: 197. https://doi.org/10.3390/genes15020197
APA StyleCornice, J., Verzella, D., Arboretto, P., Vecchiotti, D., Capece, D., Zazzeroni, F., & Franzoso, G. (2024). NF-?B: Governing Macrophages in Cancer. Genes, 15(2), 197. https://doi.org/10.3390/genes15020197