
Genetic Basis of Breast and Ovarian Cancer: Approaches and Lessons Learnt from Three Decades of Inherited Predisposition Testing

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. High-Penetrance Genes: BRCA1 and BRCA2
2.1. Pathogenic Mechanism
2.2. Types of Variants
2.3. Genotype–Phenotype Correlations
3. Other High-Penetrance Genes
3.1. CDH1
3.2. PALB2
3.3. PTEN
3.4. TP53
4. Intermediate Penetrance Genes
4.1. ATM
4.2. BARD1
4.3. BRIP1
4.4. CHEK2
4.5. RAD51C and RAD51D
5. Variants of Uncertain Significance
Functional Assays of Specific HRR Genes (Table 2)
- -
- Ubiquitin ligase activity and protein interaction: this combines ubiquitin ligase activity and yeast two-hybrid assays to assess the variant impact on the BRCA1 RING domain in mediating interactions.
- -
- Transcription Activation (TA) assay: a quantitative assay that measures the impact of variants on transactivation by the acidic C-terminal region of BRCA1 on reporter genes.
- -
- Protease sensitivity assay: this can be used to detect VUSs that affect protein folding.
- -
- Phosphopeptide binding assays: this can be used to study the interaction of BRCA1 BRCT domains with phosphorylated peptides.
- -
- Small-Colony Phenotype (SCP) assay: this reveals how BRCA1 expression impacts on yeast growth.
- -
- Yeast Localization Phenotype (YLP) assay: this can be used to investigate the cellular localization of BRCA1 in yeast.
- -
- ESC-based functional assay: this can be used to study the impact of VUSs by mouse embryonic stem cells.
- -
- Restoration of radiation resistance: this can be used to investigate if BRCA1 variants are able to restore radiation resistance.
- -
- Homology-Directed Recombination (HDR) assay: this can be used to evaluate how VUSs impact on the correct functionality of the Homologous Recombination Repair (HRR) pathway.
- -
- Centrosome amplification: this can be used to study how VUSs impact on centrosome amplification.
- -
- Yeast recombination assay: by studying the yeast HRR pathway, this can be used to evaluate the effect of BRCA1 missense VUSs.
- -
- Subcellular localization assay: this can be used to observe how BRCA1 subcellular localization varies under the influence of VUS.
- -
- Homology-Directed Recombination (HDR) assay: see previous paragraph.
- -
- HRR assay in human cells: this can be used to evaluate the effect of transient overexpression of BRCA2 VUSs on a recombination reporter substrate in HeLa G1 cells. As certain pathogenic variants exhibited effects similar to non-pathogenic ones, its specificity is uncertain.
- -
- Yeast recombination assay: human full-length BRCA2 is expressed in the yeast strain to measure HRR. HRR is increased by neutral variants, while it is decreased/is stable by pathogenic variants.
- -
- Centrosome-amplification assay: as pathogenic BRCA2 mutations induce increased centrosomes, while neutral variants and WT do not, this exhibits high specificity and reasonable sensitivity.
- -
- Mitomycin C (MMC) survival assay: this can be used to evaluate the activity of MMC on cells harboring BRCA2 VUSs (cell lines with pathogenic mutations are more sensitive to MMC).
- -
- Embryonic stem cell (ESC)-based functional assay: this can be used to study the ability of human BRCA2 VUSs to rescue ES cell viability.
- -
- Syngeneic human cancer BRCA2 knockout cell line model (SyVal model): this can be used to evaluate RAD51 foci formation and sensitivity to DNA-damaging agents introducing BRCA2 VUSs into a p53-deficient human epithelial colorectal cancer cell line.
- -
- Nuclear localization assay: as pathogenic variants exhibit cytoplasmic localization, while non-pathogenic variants remain nuclear, this can be used to observe the subcellular localization of GFP-tagged BRCA2 variants to discriminate between VUSs and pathogenic mutations.
- -
- BRCA2 protein–protein-interaction-based assays: this can be used to study how BRCA2 VUSs modify the interactions with other proteins, such as PALB2.
- -
- Analysis of variants that affect RNA splicing.
- -
- Phenotype in heterozygous carriers: cells from BRCA2 heterozygous variant carriers and healthy controls seem to behave differently upon DNA damage. Available data are still not robust to routinely use this assay to validate BRCA2 VUSs.
- -
- In vitro kinase assays;
- -
- Yeast strains expressing human CHEK2;
- -
- Knockout of breast cell lines for CHEK2.
- -
- Apoptotic pathway activation upon DNA damage: this evaluates the impact on the function of TP53 variants by verifying the capacity of the protein in activating the apoptotic pathway upon DNA damage. The cell lines used to test the apoptotic response are peripheral blood lymphocytes administered with ionizing radiation.
- -
- FASAY (Functional Assay for the Separation of Alleles in Yeast): this involves testing the ability of TP53 proteins to transactivate the ADE2 gene in yeast, providing a complementary approach to the apoptotic assay [128].

{kind=link}
{kind=link}
{kind=link}
| Assay | Description | Main Findings |
|---|---|---|
| BRCA1 Functional Assays | ||
| Ubiquitin Ligase Activity and Protein Interaction | This combines ubiquitin ligase activity and yeast two-hybrid assays to assess variant impact on BRCA1 RING domain in mediating interactions. | VUSs influence on protein conformation and interactions. |
| Transcription Activation (TA) Assay | This is a quantitative assay that measures the impact of variants on transactivation by the acidic C-terminal region of BRCA1 on the reporter gene. | VUSs affect BRCA1 transcription activation. |
| Other Functional Assays | Protease sensitivity, phosphopeptide binding, SCP, YLP, ESC-based, restoration of radiation resistance, HDR, centrosome amplification, yeast recombination, subcellular localization. | VUSs impact on protein structure, cell cycle, and response to treatment. |
| BRCA2 Functional Assays | ||
| Homology-directed repair (HDR) assay | This can be used to evaluate how VUSs impact on the correct functionality of Homologous Recombination Repair (HRR) pathway. | VUSs impact on HRR pathway. |
| Homologous recombination assay in human cells | This can be used to evaluate the effect of the transient overexpression of BRCA2 VUS on a recombination reporter substrate in HeLa G1 cells. | VUSs affect intra-chromosomal recombination. |
| Yeast recombination assay | Human full-length BRCA2 is expressed in the yeast strain to measure HRR. | HRR is increased by neutral variants, while it is decreased/is stable by pathogenic variants. |
| Centrosome-amplification assay | Pathogenic BRCA2 mutations induce increased centrosomes, while neutral variants and WT do not. | It exhibits high specificity and reasonable sensitivity. |
| Mitomycin C (MMC) survival assay | This can be used to evaluate the activity of MMC on cells harboring BRCA2 VUSs. | Cell lines with pathogenic mutations are more sensitive to MMC. |
| Embryonic stem cell (ESC)-based functional assay | This can be used to study the ability of human BRCA2 VUSs to rescue ES cell viability. | High specificity. |
| Syngeneic human cancer BRCA2 knockout cell line model | This can be used to evaluate RAD51 foci formation and sensitivity to DNA-damaging agents, introducing BRCA2 VUSs into a TP53-deficient human epithelial colorectal cancer cell line. | VUSs impact on DNA damage response. |
| Nuclear localization assay | This can be used to observe the subcellular localization of GFP-tagged BRCA2 variants. | Pathogenic variants exhibit cytoplasmic localization, while non-pathogenic variants remain nuclear. |
| BRCA2 protein–protein-interaction-based assays | This can be used to study how BRCA2 VUSs modify the interactions with other proteins. | The readout is the interaction with other proteins, such as PALB2. |
| Analysis of variants that affect RNA splicing | This can be used to investigate the presence of aberrant RNA splicing. | VUSs may influence RNA splicing. |
| Phenotype in heterozygous carriers | Cells from BRCA2 heterozygous mutation carriers and healthy controls seem to behave differently upon DNA damage. | Available data are still not robust to routinely use this assay to validate BRCA2 VUSs. |
| CHEK2 Functional Assays | ||
| Various in vitro kinase assays, budding yeast strains, and mammalian cell lines | These can be used to evaluate how VUSs impact on protein structure and activity upon DNA damage. | Using these tools, researchers have been able to classify almost 179 CHEK2 VUSs as pathogenic or not. Nonetheless, a mechanistic follow-up is needed to confirm the functional evaluation. |
| TP53 Functional Assays | ||
| Apoptotic assay | This can be used to evaluate the impact on the function of TP53 variants by verifying the capacity of the protein to activate the apoptotic pathway upon DNA damage. | The cell lines used to test the apoptotic response are peripheral blood lymphocytes administered with ionizing radiation. |
| FASAY (Functional Assay for the Separation of Alleles in Yeast) | This involves testing the ability of TP53 proteins to transactivate the ADE2 gene in yeast. | This provides a complementary approach to the apoptotic assay. |
6. Mainstreaming or Direct Genetic Testing
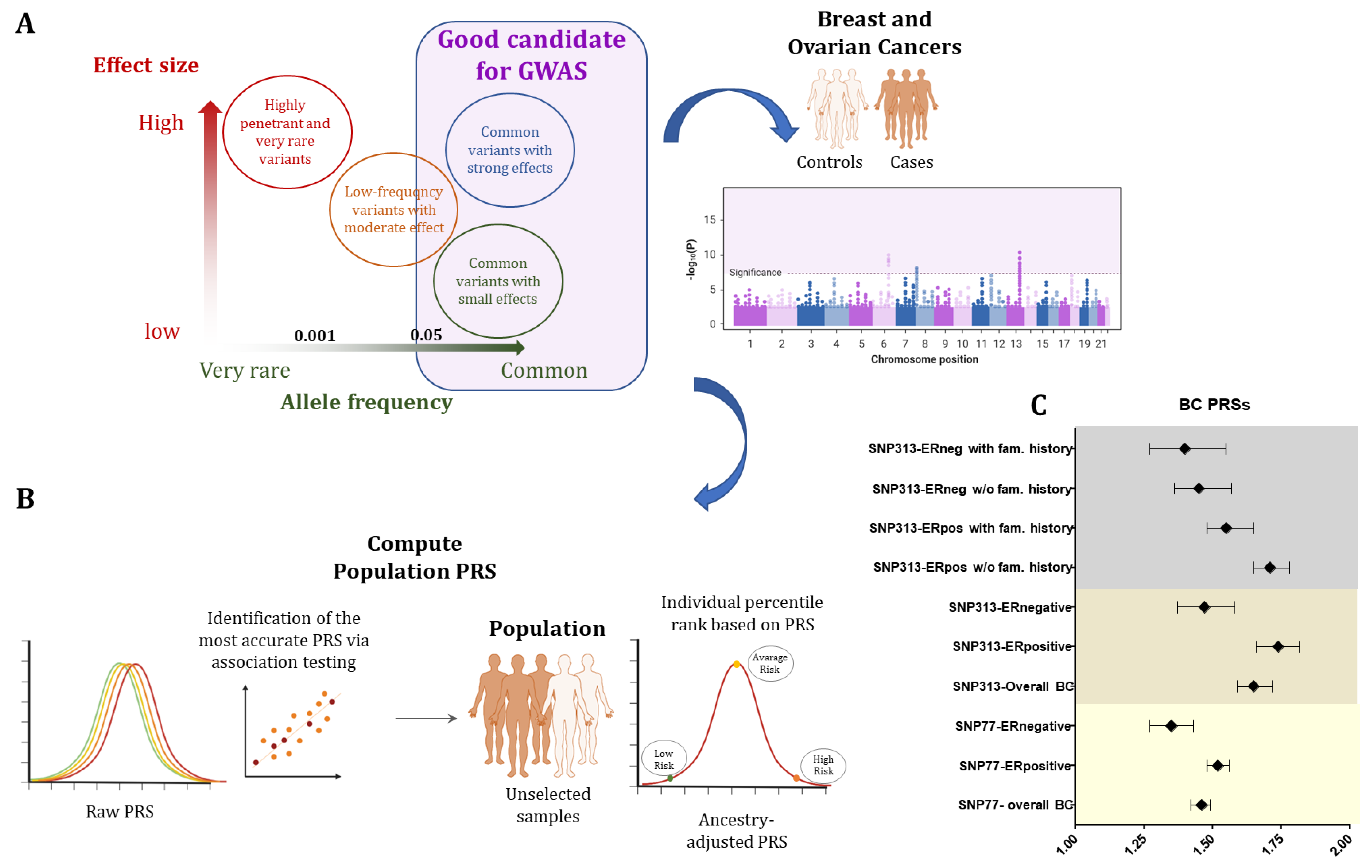
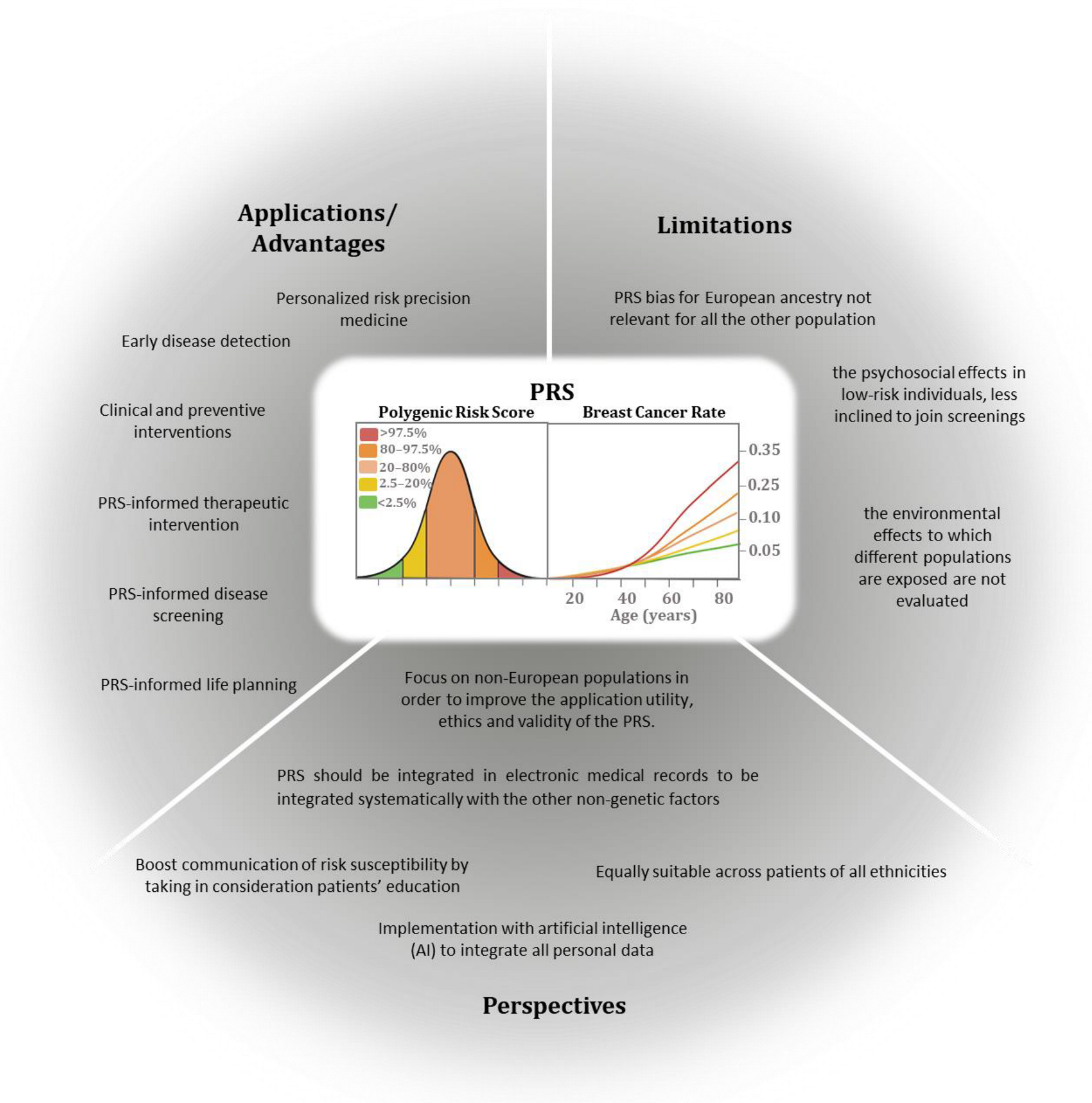
7. Polygenic Risk Score
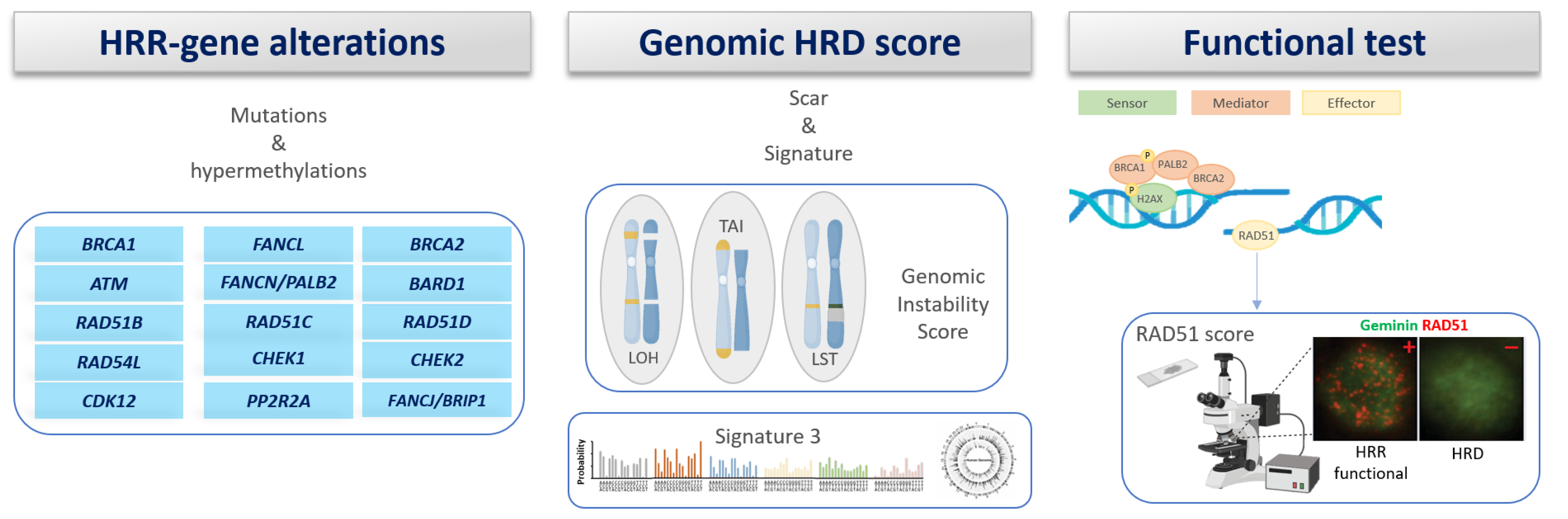
8. Homologous Recombination Deficiency (HRD)
8.1. Gene Scar/Signature
8.2. Functional Assays of HRD
8.3. RAD51 Assay as Functional Biomarker of HRD in Early BC
| Clinical Trial | Drug | Setting | Study Population | HRD Role |
|---|---|---|---|---|
| ARIEL-2 | Rucaparib | Monotherapy | Relapsed, platinum-sensitive ovarian cancer | Higher performance in gBRCA1/2-variant-associated and/or LOH-high compared to LOH-low tumors. Not effective in manifesting a difference between LOH-high and LOH-low tumors |
| ARIEL-3 | Rucaparib | Maintenance therapy | Platinum-sensitive ovarian cancer | Efficacy regardless of LOH status. |
| NOVA-trial | Niraparib | Maintenance therapy | Platinum-sensitive ovarian cancer | Efficacy regardless of HRD status. |
| Clinical Trial | Drug | Setting | Study Population | HRD Role |
|---|---|---|---|---|
| PrECOG 0105 Cisplatin-1 trial Cisplatin-2 trial | Platinum salts | Neoadjuvant setting | Untreated patients | HRD-positive patients had higher complete pathologic response |
| Gepar-Sixto trial | Carboplatin | Neoadjuvant setting | Untreated patients | HRD-positive patients have a better prognosis by comparing with HRD-negative ones. No robust evidence can be reached about the predictive role of HRD regarding carboplatin |
| TBCRC009 trial | Platinum salts | Advanced setting | First- or second-line treatment | Higher HRD scores were reported in responding patients, independent of BRCA1/2 variant status. |
| TNT trial | Carboplatinum | Advanced setting | First-line treatment | ORR is not associated with HRD levels in the primary tumors. |
9. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Broca, P. Traité des Tumeurs; P. Asselin: Paris, France, 1866. [Google Scholar]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A Strong Candidate for the Breast and Ovarian Cancer Susceptibility Gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a Breast Cancer Susceptibility Gene, BRCA2, to Chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef] [PubMed]
- Joly, Y.; Tonin, P.N. Social, Ethical and Legal Considerations Raised by the Discovery and Patenting of the BRCA1 and BRCA2 Genes. New Genet. Soc. 2014, 33, 167–180. [Google Scholar] [CrossRef]
- Easton, D.F.; Pharoah, P.D.P.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer; University of Washington: Seattle, WA, USA, 1998. [Google Scholar]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Peock, S.; Frost, D.; Ellis, S.; Platte, R.; Fineberg, E.; Evans, D.G.; Izatt, L.; Eeles, R.A.; Adlard, J.; et al. Cancer Risks for BRCA1 and BRCA2 Mutation Carriers: Results from Prospective Analysis of EMBRACE. JNCI J. Natl. Cancer Inst. 2013, 105, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, A.S.; Gong, G.; John, E.M.; McGuire, V.; Li, F.P.; Ostrow, K.L.; Dicioccio, R.; Felberg, A.; West, D.W. Prevalence of BRCA1 Mutation Carriers among U.S. Non-Hispanic Whites. Cancer Epidemiol. Biomark. Prev. 2004, 13, 2078–2083. [Google Scholar] [CrossRef]
- Manchanda, R.; Legood, R.; Burnell, M.; McGuire, A.; Raikou, M.; Loggenberg, K.; Wardle, J.; Sanderson, S.; Gessler, S.; Side, L.; et al. Cost-Effectiveness of Population Screening for BRCA Mutations in Ashkenazi Jewish Women Compared with Family History–Based Testing. JNCI J. Natl. Cancer Inst. 2015, 107, 380. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Cancer Suppression by the Chromosome Custodians, BRCA1 and BRCA2. Science 2014, 343, 1470–1475. [Google Scholar] [CrossRef]
- Sasanuma, H.; Tsuda, M.; Morimoto, S.; Saha, L.K.; Rahman, M.M.; Kiyooka, Y.; Fujiike, H.; Cherniack, A.D.; Itou, J.; Callen Moreu, E.; et al. BRCA1 Ensures Genome Integrity by Eliminating Estrogen-Induced Pathological Topoisomerase II–DNA Complexes. Proc. Natl. Acad. Sci. USA 2018, 115, E10642–E10651. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “BRCAness” in Sporadic Cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Toss, A.; Molinaro, E.; Venturelli, M.; Domati, F.; Marcheselli, L.; Piana, S.; Barbieri, E.; Grandi, G.; Piombino, C.; Marchi, I.; et al. BRCA Detection Rate in an Italian Cohort of Luminal Early-Onset and Triple-Negative Breast Cancer Patients without Family History: When Biology Overcomes Genealogy. Cancers 2020, 12, 1252. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Nepomuceno, T.C.; Foo, T.K.; Richardson, M.E.; Ranola, J.M.O.; Weyandt, J.; Varga, M.J.; Alarcon, A.; Gutierrez, D.; von Wachenfeldt, A.; Eriksson, D.; et al. BRCA1 Frameshift Variants Leading to Extended Incorrect Protein C Termini. Hum. Genet. Genom. Adv. 2023, 4, 100240. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Feroce, I.; Intra, M.; Toesca, A.; Magnoni, F.; Sargenti, M.; Naninato, P.; Caldarella, P.; Pagani, G.; Vento, A.; et al. BRCA1/2 Germline Missense Mutations: A Systematic Review. Eur. J. Cancer Prev. 2018, 27, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. How Do Mutations Affecting the Breast Cancer Genes BRCA1 and BRCA2 Cause Cancer Susceptibility? DNA Repair 2019, 81, 102668. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.R.; van Veen, E.M.; Byers, H.J.; Wallace, A.J.; Ellingford, J.M.; Beaman, G.; Santoyo-Lopez, J.; Aitman, T.J.; Eccles, D.M.; Lalloo, F.I.; et al. A Dominantly Inherited 5′ UTR Variant Causing Methylation-Associated Silencing of BRCA1 as a Cause of Breast and Ovarian Cancer. Am. J. Hum. Genet. 2018, 103, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of Type and Location of BRCA1 and BRCA2 Mutations with Risk of Breast and Ovarian Cancer. JAMA 2015, 313, 1347. [Google Scholar] [CrossRef]
- Gayther, S.A.; Mangion, J.; Russell, P.; Seal, S.; Barfoot, R.; Ponder, B.A.J.; Stratton, M.R.; Easton, D. Variation of Risks of Breast and Ovarian Cancer Associated with Different Germline Mutations of the BRCA2 Gene. Nat. Genet. 1997, 15, 103–105. [Google Scholar] [CrossRef]
- Roed Nielsen, H.; Petersen, J.; Therkildsen, C.; Skytte, A.-B.; Nilbert, M. Increased Risk of Male Cancer and Identification of a Potential Prostate Cancer Cluster Region in BRCA2. Acta Oncol. 2016, 55, 38–44. [Google Scholar] [CrossRef]
- Patel, V.L.; Busch, E.L.; Friebel, T.M.; Cronin, A.; Leslie, G.; McGuffog, L.; Adlard, J.; Agata, S.; Agnarsson, B.A.; Ahmed, M.; et al. Association of Genomic Domains in BRCA1 and BRCA2 with Prostate Cancer Risk and Aggressiveness. Cancer Res. 2020, 80, 624–638. [Google Scholar] [CrossRef]
- Chian, J.S.; Xu, W.; Wang, S.M. Pancreatic Cancer Cluster Region Identified in BRCA2. J. Med. Genet. 2023, 60, 1052–1056. [Google Scholar] [CrossRef]
- de Garibay, G.R.; Fernandez-Garcia, I.; Mazoyer, S.; Leme de Calais, F.; Ameri, P.; Vijayakumar, S.; Martinez-Ruiz, H.; Damiola, F.; Barjhoux, L.; Thomassen, M.; et al. Altered Regulation of BRCA1 Exon 11 Splicing Is Associated with Breast Cancer Risk in Carriers of BRCA1 Pathogenic Variants. Hum. Mutat. 2021, 42, 1488–1502. [Google Scholar] [CrossRef] [PubMed]
- Meeks, H.D.; Song, H.; Michailidou, K.; Bolla, M.K.; Dennis, J.; Wang, Q.; Barrowdale, D.; Frost, D.; McGuffog, L.; Ellis, S.; et al. BRCA2 Polymorphic Stop Codon K3326X and the Risk of Breast, Prostate, and Ovarian Cancers. J. Natl. Cancer Inst. 2016, 108, djv315. [Google Scholar] [CrossRef]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a Global Resource for Variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef] [PubMed]
- LaDuca, H.; Stuenkel, A.J.; Dolinsky, J.S.; Keiles, S.; Tandy, S.; Pesaran, T.; Chen, E.; Gau, C.-L.; Palmaer, E.; Shoaepour, K.; et al. Utilization of Multigene Panels in Hereditary Cancer Predisposition Testing: Analysis of More than 2,000 Patients. Genet. Med. 2014, 16, 830–837. [Google Scholar] [CrossRef]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A Study of over 35,000 Women with Breast Cancer Tested with a 25-gene Panel of Hereditary Cancer Genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Meng, H.; Yao, L.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; et al. Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin. Cancer Res. 2017, 23, 6113–6119. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Breast Cancer Association, C.; Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]
- Narod, S.A. Which Genes for Hereditary Breast Cancer? N. Engl. J. Med. 2021, 384, 471–473. [Google Scholar] [CrossRef]
- Figueiredo, J.; Melo, S.; Carneiro, P.; Moreira, A.M.; Fernandes, M.S.; Ribeiro, A.S.; Guilford, P.; Paredes, J.; Seruca, R. Clinical Spectrum and Pleiotropic Nature of CDH1 Germline Mutations. J. Med. Genet. 2019, 56, 199–208. [Google Scholar] [CrossRef]
- Kaurah, P.; MacMillan, A.; Boyd, N.; Senz, J.; De Luca, A.; Chun, N.; Suriano, G.; Zaor, S.; Van Manen, L.; Gilpin, C.; et al. Founder and Recurrent CDH1 Mutations in Families with Hereditary Diffuse Gastric Cancer. JAMA 2007, 297, 2360. [Google Scholar] [CrossRef] [PubMed]
- Benusiglio, P.R.; Malka, D.; Rouleau, E.; De Pauw, A.; Buecher, B.; Noguès, C.; Fourme, E.; Colas, C.; Coulet, F.; Warcoin, M.; et al. CDH1 Germline Mutations and the Hereditary Diffuse Gastric and Lobular Breast Cancer Syndrome: A Multicentre Study. J. Med. Genet. 2013, 50, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Blair, V.R.; McLeod, M.; Carneiro, F.; Coit, D.G.; D’Addario, J.L.; van Dieren, J.M.; Harris, K.L.; Hoogerbrugge, N.; Oliveira, C.; van der Post, R.S.; et al. Hereditary Diffuse Gastric Cancer: Updated Clinical Practice Guidelines. Lancet Oncol. 2020, 21, e386–e397. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Pinheiro, H.; Figueiredo, J.; Seruca, R.; Carneiro, F. E-Cadherin Alterations in Hereditary Disorders with Emphasis on Hereditary Diffuse Gastric Cancer. Prog. Mol. Biol. Transl. Sci. 2013, 116, 337–359. [Google Scholar] [PubMed]
- Corso, G.; Figueiredo, J.; La Vecchia, C.; Veronesi, P.; Pravettoni, G.; Macis, D.; Karam, R.; Lo Gullo, R.; Provenzano, E.; Toesca, A.; et al. Hereditary Lobular Breast Cancer with an Emphasis on E-Cadherin Genetic Defect. J. Med. Genet. 2018, 55, 431–441. [Google Scholar] [CrossRef]
- Fairoosa, P.; Witharana, C. Gene Mutations in Hereditary Breast Cancer—A Review. Eur. J. Med. Health Sci. 2020, 2. [Google Scholar] [CrossRef]
- Daly, M.B.; Pal, T.; Maxwell, K.N.; Churpek, J.; Kohlmann, W.; AlHilli, Z.; Arun, B.; Buys, S.S.; Cheng, H.; Domchek, S.M.; et al. NCCN Guidelines® Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2024. J. Natl. Compr. Canc. Netw. 2023, 21, 1000–1010. [Google Scholar] [CrossRef]
- Angeli, D.; Salvi, S.; Tedaldi, G. Genetic Predisposition to Breast and Ovarian Cancers: How Many and Which Genes to Test? Int. J. Mol. Sci. 2020, 21, 1128. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated with Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Janssen, B.; Bellis, S.; Koller, T.; Tischkowitz, M.; Liau, S.-S. A Systematic Review of Predicted Pathogenic PALB2 Variants: An Analysis of Mutational Overlap between Epithelial Cancers. J. Hum. Genet. 2020, 65, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Piombino, C.; Cortesi, L.; Lambertini, M.; Punie, K.; Grandi, G.; Toss, A. Secondary Prevention in Hereditary Breast and/or Ovarian Cancer Syndromes Other Than BRCA. J. Oncol. 2020, 2020, 6384190. [Google Scholar] [CrossRef] [PubMed]
- Nelen, M.R.; Kremer, H.; Konings, I.B.; Schoute, F.; van Essen, A.J.; Koch, R.; Woods, C.G.; Fryns, J.-P.; Hamel, B.; Hoefsloot, L.H.; et al. Novel PTEN Mutations in Patients with Cowden Disease: Absence of Clear Genotype–Phenotype Correlations. Eur. J. Hum. Genet. 1999, 7, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, R. PTEN Hamartoma Tumor Syndrome: A Clinical Overview. Cancers 2019, 11, 844. [Google Scholar] [CrossRef]
- Hendricks, L.A.J.; Hoogerbrugge, N.; Schuurs-Hoeijmakers, J.H.M.; Vos, J.R. A Review on Age-related Cancer Risks in PTEN Hamartoma Tumor Syndrome. Clin. Genet. 2021, 99, 219–225. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Colas, C.; Pouwels, S.; Hoogerbrugge, N.; Bisseling, T.; Bubien, V.; Caux, F.; Chabbert-Buffet, N.; Colas, C.; Da Mota Gomes, S.; et al. Cancer Surveillance Guideline for Individuals with PTEN Hamartoma Tumour Syndrome. Eur. J. Hum. Genet. 2020, 28, 1387–1393. [Google Scholar] [CrossRef]
- Hendricks, L.A.J.; Hoogerbrugge, N.; Mensenkamp, A.R.; Brunet, J.; Lleuger-Pujol, R.; Høberg-Vetti, H.; Tveit Haavind, M.; Innella, G.; Turchetti, D.; Aretz, S.; et al. Cancer Risks by Sex and Variant Type in PTEN Hamartoma Tumor Syndrome. JNCI J. Natl. Cancer Inst. 2023, 115, 93–103. [Google Scholar] [CrossRef]
- Frazier, T.W.; Embacher, R.; Tilot, A.K.; Koenig, K.; Mester, J.; Eng, C. Molecular and Phenotypic Abnormalities in Individuals with Germline Heterozygous PTEN Mutations and Autism. Mol. Psychiatry 2015, 20, 1132–1138. [Google Scholar] [CrossRef]
- Gonzalez, K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Wen-Fong, C.Y.; Nguyen, V.Q.; Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li Fraumeni Syndrome: Clinical Characteristics of Families with P53 Germline Mutations. J. Clin. Oncol. 2009, 27, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Mai, P.L.; Best, A.F.; Peters, J.A.; DeCastro, R.M.; Khincha, P.P.; Loud, J.T.; Bremer, R.C.; Rosenberg, P.S.; Savage, S.A. Risks of First and Subsequent Cancers among TP53 Mutation Carriers in the National Cancer Institute Li-Fraumeni Syndrome Cohort. Cancer 2016, 122, 3673–3681. [Google Scholar] [CrossRef] [PubMed]
- Melhem-Bertrandt, A.; Bojadzieva, J.; Ready, K.J.; Obeid, E.; Liu, D.D.; Gutierrez-Barrera, A.M.; Litton, J.K.; Olopade, O.I.; Hortobagyi, G.N.; Strong, L.C.; et al. Early Onset HER2-Positive Breast Cancer Is Associated with Germline TP53 Mutations. Cancer 2012, 118, 908–913. [Google Scholar] [CrossRef]
- Frebourg, T.; Bajalica Lagercrantz, S.; Oliveira, C.; Magenheim, R.; Evans, D.G. Guidelines for the Li–Fraumeni and Heritable TP53-Related Cancer Syndromes. Eur. J. Hum. Genet. 2020, 28, 1379–1386. [Google Scholar] [CrossRef]
- Renaux-Petel, M.; Charbonnier, F.; Théry, J.-C.; Fermey, P.; Lienard, G.; Bou, J.; Coutant, S.; Vezain, M.; Kasper, E.; Fourneaux, S.; et al. Contribution of de Novo and Mosaic TP53 Mutations to Li-Fraumeni Syndrome. J. Med. Genet. 2018, 55, 173–180. [Google Scholar] [CrossRef]
- Weitzel, J.N.; Chao, E.C.; Nehoray, B.; Van Tongeren, L.R.; LaDuca, H.; Blazer, K.R.; Slavin, T.; Pesaran, T.; Rybak, C.; Solomon, I.; et al. Somatic TP53 Variants Frequently Confound Germ-Line Testing Results. Genet. Med. 2018, 20, 809–816. [Google Scholar] [CrossRef]
- Chen, S.; Liu, Y. P53 Involvement in Clonal Hematopoiesis of Indeterminate Potential. Curr. Opin. Hematol. 2019, 26, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Achatz, M.I.W.; Olivier, M.; Le Calvez, F.; Martel-Planche, G.; Lopes, A.; Rossi, B.M.; Ashton-Prolla, P.; Giugliani, R.; Palmero, E.I.; Vargas, F.R.; et al. The TP53 Mutation, R337H, Is Associated with Li-Fraumeni and Li-Fraumeni-like Syndromes in Brazilian Families. Cancer Lett. 2007, 245, 96–102. [Google Scholar] [CrossRef]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef]
- Swift, M.; Morrell, D.; Cromartie, E.; Chamberlin, A.R.; Skolnick, M.H.; Bishop, D.T. The Incidence and Gene Frequency of Ataxia-Telangiectasia in the United States. Am. J. Hum. Genet. 1986, 39, 573–583. [Google Scholar]
- Lu, H.-M.; Li, S.; Black, M.H.; Lee, S.; Hoiness, R.; Wu, S.; Mu, W.; Huether, R.; Chen, J.; Sridhar, S.; et al. Association of Breast and Ovarian Cancers with Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol. 2019, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM Mutations in Patients with Hereditary Pancreatic Cancer. Cancer Discov. 2012, 2, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Helgason, H.; Rafnar, T.; Olafsdottir, H.S.; Jonasson, J.G.; Sigurdsson, A.; Stacey, S.N.; Jonasdottir, A.; Tryggvadottir, L.; Alexiusdottir, K.; Haraldsson, A.; et al. Loss-of-Function Variants in ATM Confer Risk of Gastric Cancer. Nat. Genet. 2015, 47, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.L.; Concannon, P. ATM, Radiation, and the Risk of Second Primary Breast Cancer. Int. J. Radiat. Biol. 2017, 93, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Goldgar, D.E.; Healey, S.; Dowty, J.G.; Da Silva, L.; Chen, X.; Spurdle, A.B.; Terry, M.B.; Daly, M.J.; Buys, S.M.; Southey, M.C.; et al. Rare Variants in the ATMgene and Risk of Breast Cancer. Breast Cancer Res. 2011, 13, R73. [Google Scholar] [CrossRef]
- Hall, M.J.; Bernhisel, R.; Hughes, E.; Larson, K.; Rosenthal, E.T.; Singh, N.A.; Lancaster, J.M.; Kurian, A.W. Germline Pathogenic Variants in the Ataxia Telangiectasia Mutated (ATM) Gene Are Associated with High and Moderate Risks for Multiple Cancers. Cancer Prev. Res. 2021, 14, 433–440. [Google Scholar] [CrossRef]
- Tung, N.M.; Boughey, J.C.; Pierce, L.J.; Robson, M.E.; Bedrosian, I.; Dietz, J.R.; Dragun, A.; Gelpi, J.B.; Hofstatter, E.W.; Isaacs, C.J.; et al. Management of Hereditary Breast Cancer: American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology Guideline. J. Clin. Oncol. 2020, 38, 2080–2106. [Google Scholar] [CrossRef]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.-C.W.; Hwang, L.-Y.; Bowcock, A.M.; Baer, R. Identification of a RING Protein That Can Interact in Vivo with the BRCA1 Gene Product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Śniadecki, M.; Brzeziński, M.; Darecka, K.; Klasa-Mazurkiewicz, D.; Poniewierza, P.; Krzeszowiec, M.; Kmieć, N.; Wydra, D. BARD1 and Breast Cancer: The Possibility of Creating Screening Tests and New Preventive and Therapeutic Pathways for Predisposed Women. Genes 2020, 11, 1251. [Google Scholar] [CrossRef]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190. [Google Scholar] [CrossRef] [PubMed]
- Shimelis, H.; LaDuca, H.; Hu, C.; Hart, S.N.; Na, J.; Thomas, A.; Akinhanmi, M.; Moore, R.M.; Brauch, H.; Cox, A.; et al. Triple-Negative Breast Cancer Risk Genes Identified by Multigene Hereditary Cancer Panel Testing. JNCI J. Natl. Cancer Inst. 2018, 110, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Devoto, M.; Hou, C.; Asgharzadeh, S.; Glessner, J.T.; Attiyeh, E.F.; Mosse, Y.P.; Kim, C.; Diskin, S.J.; Cole, K.A.; et al. Common Variations in BARD1 Influence Susceptibility to High-Risk Neuroblastoma. Nat. Genet. 2009, 41, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Weber-Lassalle, N.; Borde, J.; Weber-Lassalle, K.; Horváth, J.; Niederacher, D.; Arnold, N.; Kaulfuß, S.; Ernst, C.; Paul, V.G.; Honisch, E.; et al. Germline Loss-of-Function Variants in the BARD1 Gene Are Associated with Early-Onset Familial Breast Cancer but Not Ovarian Cancer. Breast Cancer Res. 2019, 21, 55. [Google Scholar] [CrossRef]
- Nalepa, G.; Clapp, D.W. Fanconi Anaemia and Cancer: An Intricate Relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef]
- Seal, S.; Thompson, D.; Renwick, A.; Elliott, A.; Kelly, P.; Barfoot, R.; Chagtai, T.; Jayatilake, H.; Ahmed, M.; Spanova, K.; et al. Truncating Mutations in the Fanconi Anemia J Gene BRIP1 Are Low-Penetrance Breast Cancer Susceptibility Alleles. Nat. Genet. 2006, 38, 1239–1241. [Google Scholar] [CrossRef]
- Easton, D.F.; Lesueur, F.; Decker, B.; Michailidou, K.; Li, J.; Allen, J.; Luccarini, C.; Pooley, K.A.; Shah, M.; Bolla, M.K.; et al. No Evidence That Protein Truncating Variants in BRIP1 Are Associated with Breast Cancer Risk: Implications for Gene Panel Testing. J. Med. Genet. 2016, 53, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D Mutations Are Associated with High Susceptibility to Ovarian Cancer: Mutation Prevalence and Precise Risk Estimates Based on a Pooled Analysis of ~30,000 Cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Moyer, C.L.; Ivanovich, J.; Gillespie, J.L.; Doberstein, R.; Radke, M.R.; Richardson, M.E.; Kaufmann, S.H.; Swisher, E.M.; Goodfellow, P.J. Rare BRIP1 Missense Alleles Confer Risk for Ovarian and Breast Cancer. Cancer Res. 2020, 80, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Wokołorczyk, D.; Jakubowska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Masojć, B.; Dębniak, T.; Górski, B.; Blecharz, P.; et al. Risk of Breast Cancer in Women with a CHEK2 Mutation with and without a Family History of Breast Cancer. J. Clin. Oncol. 2011, 29, 3747–3752. [Google Scholar] [CrossRef] [PubMed]
- Adank, M.A.; Jonker, M.A.; Kluijt, I.; van Mil, S.E.; Oldenburg, R.A.; Mooi, W.J.; Hogervorst, F.B.L.; van den Ouweland, A.M.W.; Gille, J.J.P.; Schmidt, M.K.; et al. CHEK2*1100delC Homozygosity Is Associated with a High Breast Cancer Risk in Women. J. Med. Genet. 2011, 48, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Kilpivaara, O.; Vahteristo, P.; Falck, J.; Syrjäkoski, K.; Eerola, H.; Easton, D.; Bartkova, J.; Lukas, J.; Heikkilä, P.; Aittomäki, K.; et al. CHEK2 Variant I157T May Be Associated with Increased Breast Cancer Risk. Int. J. Cancer 2004, 111, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Decker, B.; Allen, J.; Luccarini, C.; Pooley, K.A.; Shah, M.; Bolla, M.K.; Wang, Q.; Ahmed, S.; Baynes, C.; Conroy, D.M.; et al. Rare, Protein-Truncating Variants in ATM, CHEK2 and PALB2, but Not XRCC2, Are Associated with Increased Breast Cancer Risks. J. Med. Genet. 2017, 54, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Petridis, C.; Arora, I.; Shah, V.; Moss, C.L.; Mera, A.; Clifford, A.; Gillett, C.; Pinder, S.E.; Tomlinson, I.; Roylance, R.; et al. Frequency of Pathogenic Germline Variants in CDH1, BRCA2, CHEK2, PALB2, BRCA1, and TP53 in Sporadic Lobular Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1162–1168. [Google Scholar] [CrossRef]
- Muranen, T.A.; Greco, D.; Blomqvist, C.; Aittomäki, K.; Khan, S.; Hogervorst, F.; Verhoef, S.; Pharoah, P.D.P.; Dunning, A.M.; Shah, M.; et al. Genetic Modifiers of CHEK2*1100delC-Associated Breast Cancer Risk. Genet. Med. 2017, 19, 599–603. [Google Scholar] [CrossRef]
- Katona, B.W.; Yurgelun, M.B.; Garber, J.E.; Offit, K.; Domchek, S.M.; Robson, M.E.; Stadler, Z.K. A Counseling Framework for Moderate-Penetrance Colorectal Cancer Susceptibility Genes. Genet. Med. 2018, 20, 1324–1327. [Google Scholar] [CrossRef]
- Carlo, M.I.; Mukherjee, S.; Mandelker, D.; Vijai, J.; Kemel, Y.; Zhang, L.; Knezevic, A.; Patil, S.; Ceyhan-Birsoy, O.; Huang, K.-C.; et al. Prevalence of Germline Mutations in Cancer Susceptibility Genes in Patients with Advanced Renal Cell Carcinoma. JAMA Oncol. 2018, 4, 1228. [Google Scholar] [CrossRef]
- Siołek, M.; Cybulski, C.; Gąsior-Perczak, D.; Kowalik, A.; Kozak-Klonowska, B.; Kowalska, A.; Chłopek, M.; Kluźniak, W.; Wokołorczyk, D.; Pałyga, I.; et al. CHEK2 Mutations and the Risk of Papillary Thyroid Cancer. Int. J. Cancer 2015, 137, 548–552. [Google Scholar] [CrossRef]
- AlDubayan, S.H.; Pyle, L.C.; Gamulin, M.; Kulis, T.; Moore, N.D.; Taylor-Weiner, A.; Hamid, A.A.; Reardon, B.; Wubbenhorst, B.; Godse, R.; et al. Association of Inherited Pathogenic Variants in Checkpoint Kinase 2 (CHEK2) with Susceptibility to Testicular Germ Cell Tumors. JAMA Oncol. 2019, 5, 514. [Google Scholar] [CrossRef] [PubMed]
- Pritzlaff, M.; Summerour, P.; McFarland, R.; Li, S.; Reineke, P.; Dolinsky, J.S.; Goldgar, D.E.; Shimelis, H.; Couch, F.J.; Chao, E.C.; et al. Male Breast Cancer in a Multi-Gene Panel Testing Cohort: Insights and Unexpected Results. Breast Cancer Res. Treat. 2017, 161, 575–586. [Google Scholar] [CrossRef] [PubMed]
- LaDuca, H.; Polley, E.C.; Yussuf, A.; Hoang, L.; Gutierrez, S.; Hart, S.N.; Yadav, S.; Hu, C.; Na, J.; Goldgar, D.E.; et al. A Clinical Guide to Hereditary Cancer Panel Testing: Evaluation of Gene-Specific Cancer Associations and Sensitivity of Genetic Testing Criteria in a Cohort of 165,000 High-Risk Patients. Genet. Med. 2020, 22, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Szymanska-Pasternak, J.; Szymanska, A.; Medrek, K.; Imyanitov, E.N.; Cybulski, C.; Gorski, B.; Magnowski, P.; Dziuba, I.; Gugala, K.; Debniak, B.; et al. CHEK2 Variants Predispose to Benign, Borderline and Low-Grade Invasive Ovarian Tumors. Gynecol. Oncol. 2006, 102, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Pelttari, L.M.; Kiiski, J.; Nurminen, R.; Kallioniemi, A.; Schleutker, J.; Gylfe, A.; Aaltonen, L.A.; Leminen, A.; Heikkilä, P.; Blomqvist, C.; et al. A Finnish Founder Mutation in RAD51D: Analysis in Breast, Ovarian, Prostate, and Colorectal Cancer: Table 1. J. Med. Genet. 2012, 49, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Xie, Y. Associations between RAD51D Germline Mutations and Breast Cancer Risk and Survival in BRCA1/2-Negative Breast Cancers. Ann. Oncol. 2018, 29, 2046–2051. [Google Scholar] [CrossRef]
- Ricker, C.; Culver, J.O.; Lowstuter, K.; Sturgeon, D.; Sturgeon, J.D.; Chanock, C.R.; Gauderman, W.J.; McDonnell, K.J.; Idos, G.E.; Gruber, S.B. Increased Yield of Actionable Mutations Using Multi-Gene Panels to Assess Hereditary Cancer Susceptibility in an Ethnically Diverse Clinical Cohort. Cancer Genet. 2016, 209, 130–137. [Google Scholar] [CrossRef]
- Kurian, A.W.; Hare, E.E.; Mills, M.A.; Kingham, K.E.; McPherson, L.; Whittemore, A.S.; McGuire, V.; Ladabaum, U.; Kobayashi, Y.; Lincoln, S.E.; et al. Clinical Evaluation of a Multiple-Gene Sequencing Panel for Hereditary Cancer Risk Assessment. J. Clin. Oncol. 2014, 32, 2001–2009. [Google Scholar] [CrossRef]
- Brnich, S.E.; Abou Tayoun, A.N.; Couch, F.J.; Cutting, G.R.; Greenblatt, M.S.; Heinen, C.D.; Kanavy, D.M.; Luo, X.; McNulty, S.M.; Starita, L.M.; et al. Recommendations for Application of the Functional Evidence PS3/BS3 Criterion Using the ACMG/AMP Sequence Variant Interpretation Framework. Genome Med. 2020, 12, 3. [Google Scholar] [CrossRef]
- Ng, P.C. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Röner, S.; Mai, K.; Klinkhammer, H.; Kircher, M.; Ludwig, K.U. Predicting the Pathogenicity of Missense Variants Using Features Derived from AlphaFold2. Bioinformatics 2023, 39, btad280. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate Proteome-Wide Missense Variant Effect Prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Starita, L.M.; Ahituv, N.; Dunham, M.J.; Kitzman, J.O.; Roth, F.P.; Seelig, G.; Shendure, J.; Fowler, D.M. Variant Interpretation: Functional Assays to the Rescue. Am. J. Hum. Genet. 2017, 101, 315–325. [Google Scholar] [CrossRef]
- Findlay, G.M.; Boyle, E.A.; Hause, R.J.; Klein, J.C.; Shendure, J. Saturation Editing of Genomic Regions by Multiplex Homology-Directed Repair. Nature 2014, 513, 120–123. [Google Scholar] [CrossRef]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate Classification of BRCA1 Variants with Saturation Genome Editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef]
- Sahu, S.; Sullivan, T.L.; Mitrophanov, A.Y.; Galloux, M.; Nousome, D.; Southon, E.; Caylor, D.; Mishra, A.P.; Evans, C.N.; Clapp, M.E.; et al. Saturation Genome Editing of 11 Codons and Exon 13 of BRCA2 Coupled with Chemotherapeutic Drug Response Accurately Determines Pathogenicity of Variants. PLoS Genet. 2023, 19, e1010940. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Hu, C.; Na, J.; Hart, S.N.; David Gnanaolivu, R.; Abozaid, M.; Rao, T.; Tecleab, Y.A.; Pesaran, T.; Cilas Morais Lyra, P.; et al. Saturation Genome Editing-Based Functional Evaluation and Clinical 2 Classification of BRCA2 Single Nucleotide Variants. bioRxiv 2023. [Google Scholar] [CrossRef]
- Fayer, S.; Horton, C.; Dines, J.N.; Rubin, A.F.; Richardson, M.E.; McGoldrick, K.; Hernandez, F.; Pesaran, T.; Karam, R.; Shirts, B.H.; et al. Closing the Gap: Systematic Integration of Multiplexed Functional Data Resolves Variants of Uncertain Significance in BRCA1, TP53, and PTEN. Am. J. Hum. Genet. 2021, 108, 2248–2258. [Google Scholar] [CrossRef] [PubMed]
- Esposito, D.; Weile, J.; Shendure, J.; Starita, L.M.; Papenfuss, A.T.; Roth, F.P.; Fowler, D.M.; Rubin, A.F. MaveDB: An Open-Source Platform to Distribute and Interpret Data from Multiplexed Assays of Variant Effect. Genome Biol. 2019, 20, 223. [Google Scholar] [CrossRef] [PubMed]
- Boonen, R.A.C.M.; Vreeswijk, M.P.G.; van Attikum, H. Functional Characterization of PALB2 Variants of Uncertain Significance: Toward Cancer Risk and Therapy Response Prediction. Front. Mol. Biosci. 2020, 7, 169. [Google Scholar] [CrossRef] [PubMed]
- Boonen, R.A.C.M.; Wiegant, W.W.; Celosse, N.; Vroling, B.; Heijl, S.; Kote-Jarai, Z.; Mijuskovic, M.; Cristea, S.; Solleveld-Westerink, N.; van Wezel, T.; et al. Functional Analysis Identifies Damaging CHEK2 Missense Variants Associated with Increased Cancer Risk. Cancer Res. 2022, 82, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Lohmueller, K.E.; Indap, A.R.; Schmidt, S.; Boyko, A.R.; Hernandez, R.D.; Hubisz, M.J.; Sninsky, J.J.; White, T.J.; Sunyaev, S.R.; Nielsen, R.; et al. Proportionally More Deleterious Genetic Variation in European than in African Populations. Nature 2008, 451, 994–997. [Google Scholar] [CrossRef]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen—The Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef]
- Fowler, D.M.; Adams, D.J.; Gloyn, A.L.; Hahn, W.C.; Marks, D.S.; Muffley, L.A.; Neal, J.T.; Roth, F.P.; Rubin, A.F.; Starita, L.M.; et al. An Atlas of Variant Effects to Understand the Genome at Nucleotide Resolution. Genome Biol. 2023, 24, 147. [Google Scholar] [CrossRef]
- Parsons, M.T.; Tudini, E.; Li, H.; Hahnen, E.; Wappenschmidt, B.; Feliubadaló, L.; Aalfs, C.M.; Agata, S.; Aittomäki, K.; Alducci, E.; et al. Large Scale Multifactorial Likelihood Quantitative Analysis of BRCA1and BRCA2 Variants: An ENIGMA Resource to Support Clinical Variant Classification. Hum. Mutat. 2019, 40, 1557–1578. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Healey, S.; Devereau, A.; Hogervorst, F.B.L.; Monteiro, A.N.A.; Nathanson, K.L.; Radice, P.; Stoppa-Lyonnet, D.; Tavtigian, S.; Wappenschmidt, B.; et al. ENIGMA-Evidence-Based Network for the Interpretation of Germline Mutant Alleles: An International Initiative to Evaluate Risk and Clinical Significance Associated with Sequence Variation in BRCA1 and BRCA2 Genes. Hum. Mutat. 2012, 33, 2–7. [Google Scholar] [CrossRef]
- Prince, A.E.R.; Berg, J.S.; Evans, J.P.; Jonas, D.E.; Henderson, G. Genomic Screening of the General Adult Population: Key Concepts for Assessing Net Benefit with Systematic Evidence Reviews. Genet. Med. 2015, 17, 441–443. [Google Scholar] [CrossRef]
- Millot, G.A.; Carvalho, M.A.; Caputo, S.M.; Vreeswijk, M.P.G.; Brown, M.A.; Webb, M.; Rouleau, E.; Neuhausen, S.L.; Hansen, T.v.O.; Galli, A.; et al. A Guide for Functional Analysis of BRCA1 Variants of Uncertain Significance. Hum. Mutat. 2012, 33, 1526–1537. [Google Scholar] [CrossRef]
- Guidugli, L.; Carreira, A.; Caputo, S.M.; Ehlen, A.; Galli, A.; Monteiro, A.N.A.; Neuhausen, S.L.; Hansen, T.V.O.; Couch, F.J.; Vreeswijk, M.P.G. Functional Assays for Analysis of Variants of Uncertain Significance in BRCA2. Hum. Mutat. 2014, 35, 151–164. [Google Scholar] [CrossRef]
- Boonen, R.A.C.M.; Vreeswijk, M.P.G.; van Attikum, H. CHEK2 Variants: Linking Functional Impact to Cancer Risk. Trends Cancer 2022, 8, 759–770. [Google Scholar] [CrossRef]
- Camplejohn, R.S.; Rutherford, J. P53 Functional Assays: Detecting P53 Mutations in Both the Germline and in Sporadic Tumours. Cell Prolif. 2001, 34, 1–14. [Google Scholar] [CrossRef]
- McCuaig, J.; Armel, S.; Care, M.; Volenik, A.; Kim, R.; Metcalfe, K. Next-Generation Service Delivery: A Scoping Review of Patient Outcomes Associated with Alternative Models of Genetic Counseling and Genetic Testing for Hereditary Cancer. Cancers 2018, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Riddell, D.; Seal, S.; Talukdar, S.; Mahamdallie, S.; Ruark, E.; Cloke, V.; Slade, I.; Kemp, Z.; Gore, M.; et al. Implementing Rapid, Robust, Cost-Effective, Patient-Centred, Routine Genetic Testing in Ovarian Cancer Patients. Sci. Rep. 2016, 6, 29506. [Google Scholar] [CrossRef]
- Rahman, B.; Lanceley, A.; Kristeleit, R.S.; Ledermann, J.A.; Lockley, M.; McCormack, M.; Mould, T.; Side, L. Mainstreamed Genetic Testing for Women with Ovarian Cancer: First-Year Experience. J. Med. Genet. 2019, 56, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Hallowell, N.; Wright, S.; Stirling, D.; Gourley, C.; Young, O.; Porteous, M. Moving into the Mainstream: Healthcare Professionals’ Views of Implementing Treatment Focussed Genetic Testing in Breast Cancer Care. Fam. Cancer 2019, 18, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Mikat-Stevens, N.A.; Larson, I.A.; Tarini, B.A. Primary-Care Providers’ Perceived Barriers to Integration of Genetics Services: A Systematic Review of the Literature. Genet. Med. 2015, 17, 169–176. [Google Scholar] [CrossRef]
- Hamilton, J.G.; Abdiwahab, E.; Edwards, H.M.; Fang, M.-L.; Jdayani, A.; Breslau, E.S. Primary Care Providers’ Cancer Genetic Testing-Related Knowledge, Attitudes, and Communication Behaviors: A Systematic Review and Research Agenda. J. Gen. Intern. Med. 2017, 32, 315–324. [Google Scholar] [CrossRef]
- Wilkes, M.S.; Day, F.C.; Fancher, T.L.; McDermott, H.; Lehman, E.; Bell, R.A.; Green, M.J. Increasing Confidence and Changing Behaviors in Primary Care Providers Engaged in Genetic Counselling. BMC Med. Educ. 2017, 17, 163. [Google Scholar] [CrossRef]
- Scheuner, M.T.; Hamilton, A.B.; Peredo, J.; Sale, T.J.; Austin, C.; Gilman, S.C.; Bowen, M.S.; Goldzweig, C.L.; Lee, M.; Mittman, B.S.; et al. A Cancer Genetics Toolkit Improves Access to Genetic Services through Documentation and Use of the Family History by Primary-Care Clinicians. Genet. Med. 2014, 16, 60–69. [Google Scholar] [CrossRef]
- Chatterjee, N.; Shi, J.; García-Closas, M. Developing and Evaluating Polygenic Risk Prediction Models for Stratified Disease Prevention. Nat. Rev. Genet. 2016, 17, 392–406. [Google Scholar] [CrossRef]
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The Personal and Clinical Utility of Polygenic Risk Scores. Nat. Rev. Genet. 2018, 19, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Pharoah, P.D.P.; Michailidou, K.; Tyrer, J.; Brook, M.N.; Bolla, M.K.; Wang, Q.; Dennis, J.; Dunning, A.M.; Shah, M.; et al. Prediction of Breast Cancer Risk Based on Profiling with Common Genetic Variants. JNCI J. Natl. Cancer Inst. 2015, 107, djv036. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Leslie, G.; Gentry-Maharaj, A.; Ryan, A.; Intermaggio, M.; Lee, A.; Kalsi, J.K.; Tyrer, J.; Gaba, F.; Manchanda, R.; et al. Evaluation of Polygenic Risk Scores for Ovarian Cancer Risk Prediction in a Prospective Cohort Study. J. Med. Genet. 2018, 55, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Brentnall, A.; Byers, H.; Harkness, E.; Stavrinos, P.; Howell, A.; Newman, W.G.; Cuzick, J. The Impact of a Panel of 18 SNPs on Breast Cancer Risk in Women Attending a UK Familial Screening Clinic: A Case–Control Study. J. Med. Genet. 2017, 54, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.P.; Chen, T.-H.; Wang, Q.; Bolla, M.K.; et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; McGuffog, L.; Barrowdale, D.; Lee, A.; Soucy, P.; Dennis, J.; Domchek, S.M.; Robson, M.; Spurdle, A.B.; Ramus, S.J.; et al. Evaluation of Polygenic Risk Scores for Breast and Ovarian Cancer Risk Prediction in BRCA1 and BRCA2 Mutation Carriers. JNCI J. Natl. Cancer Inst. 2017, 10, djw302. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.R.; Rookus, M.A.; McGuffog, L.; Leslie, G.; Mooij, T.M.; Dennis, J.; Mavaddat, N.; Adlard, J.; Ahmed, M.; Aittomäki, K.; et al. Polygenic Risk Scores and Breast and Epithelial Ovarian Cancer Risks for Carriers of BRCA1 and BRCA2 Pathogenic Variants. Genet. Med. 2020, 22, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Amir, E.; Freedman, O.C.; Seruga, B.; Evans, D.G. Assessing Women at High Risk of Breast Cancer: A Review of Risk Assessment Models. JNCI J. Natl. Cancer Inst. 2010, 102, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Mavaddat, N.; Wilcox, A.N.; Cunningham, A.P.; Carver, T.; Hartley, S.; Babb de Villiers, C.; Izquierdo, A.; Simard, J.; Schmidt, M.K.; et al. BOADICEA: A Comprehensive Breast Cancer Risk Prediction Model Incorporating Genetic and Nongenetic Risk Factors. Genet. Med. 2019, 21, 1708–1718. [Google Scholar] [CrossRef]
- Badré, A.; Zhang, L.; Muchero, W.; Reynolds, J.C.; Pan, C. Deep Neural Network Improves the Estimation of Polygenic Risk Scores for Breast Cancer. J. Hum. Genet. 2021, 66, 359–369. [Google Scholar] [CrossRef]
- Amos, C.I.; Dennis, J.; Wang, Z.; Byun, J.; Schumacher, F.R.; Gayther, S.A.; Casey, G.; Hunter, D.J.; Sellers, T.A.; Gruber, S.B.; et al. The OncoArray Consortium: A Network for Understanding the Genetic Architecture of Common Cancers. Cancer Epidemiol. Biomark. Prev. 2017, 26, 126–135. [Google Scholar] [CrossRef]
- Mital, S.; Nguyen, H.V. Cost-Effectiveness of Using Artificial Intelligence versus Polygenic Risk Score to Guide Breast Cancer Screening. BMC Cancer 2022, 22, 501. [Google Scholar] [CrossRef]
- Areia, M.; Mori, Y.; Correale, L.; Repici, A.; Bretthauer, M.; Sharma, P.; Taveira, F.; Spadaccini, M.; Antonelli, G.; Ebigbo, A.; et al. Cost-Effectiveness of Artificial Intelligence for Screening Colonoscopy: A Modelling Study. Lancet Digit. Health 2022, 4, e436–e444. [Google Scholar] [CrossRef]
- Morrison, S.L.; Dukhovny, D.; Chan, R.V.P.; Chiang, M.F.; Campbell, J.P. Cost-Effectiveness of Artificial Intelligence–Based Retinopathy of Prematurity Screening. JAMA Ophthalmol. 2022, 140, 401. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic Scars as Biomarkers of Homologous Recombination Deficiency and Drug Response in Breast and Ovarian Cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib Maintenance Treatment for Recurrent Ovarian Carcinoma after Response to Platinum Therapy (ARIEL3): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous Recombination DNA Repair Deficiency and PARP Inhibition Activity in Primary Triple Negative Breast Cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Garg, G.; Shah, J.P.; Kumar, S.; Bryant, C.S.; Munkarah, A.; Morris, R.T. Ovarian and Uterine Carcinosarcomas: A Comparative Analysis of Prognostic Variables and Survival Outcomes. Int. J. Gynecol. Cancer 2010, 20, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Graeser, M.; McCarthy, A.; Lord, C.J.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.S.; et al. A Marker of Homologous Recombination Predicts Pathologic Complete Response to Neoadjuvant Chemotherapy in Primary Breast Cancer. Clin. Cancer Res. 2010, 16, 6159–6168. [Google Scholar] [CrossRef] [PubMed]
- Naipal, K.A.T.; Verkaik, N.S.; Ameziane, N.; van Deurzen, C.H.M.; ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional Ex Vivo Assay to Select Homologous Recombination–Deficient Breast Tumors for PARP Inhibitor Treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 Foci as a Functional Biomarker of Homologous Recombination Repair and PARP Inhibitor Resistance in Germline BRCA-Mutated Breast Cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD51 Assay Feasible in Routine Tumor Samples Calls PARP Inhibitor Response beyond BRCA Mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Bakr, A.; Oing, C.; Köcher, S.; Borgmann, K.; Dornreiter, I.; Petersen, C.; Dikomey, E.; Mansour, W.Y. Involvement of ATM in Homologous Recombination after End Resection and RAD51 Nucleofilament Formation. Nucleic Acids Res. 2015, 43, 3154–3166. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Białkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to Poly(ADP-Ribose) Polymerase Inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed]
- Llop-Guevara, A.; Loibl, S.; Villacampa, G.; Vladimirova, V.; Schneeweiss, A.; Karn, T.; Zahm, D.-M.; Herencia-Ropero, A.; Jank, P.; van Mackelenbergh, M.; et al. Association of RAD51 with Homologous Recombination Deficiency (HRD) and Clinical Outcomes in Untreated Triple-Negative Breast Cancer (TNBC): Analysis of the GeparSixto Randomized Clinical Trial. Ann. Oncol. 2021, 32, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib Monotherapy as Primary Treatment in Unselected Triple Negative Breast Cancer. Ann. Oncol. 2021, 32, 240–249. [Google Scholar] [CrossRef]
- So, M.-K.; Jeong, T.-D.; Lim, W.; Moon, B.-I.; Paik, N.S.; Kim, S.C.; Huh, J. Reinterpretation of BRCA1 and BRCA2 Variants of Uncertain Significance in Patients with Hereditary Breast/Ovarian Cancer Using the ACMG/AMP 2015 Guidelines. Breast Cancer 2019, 26, 510–519. [Google Scholar] [CrossRef]
- Martorana, D.; Barili, V.; Uliana, V.; Ambrosini, E.; Riva, M.; De Sensi, E.; Luppi, E.; Messina, C.; Caleffi, E.; Pisani, F.; et al. Reassessment of the NF1 Variants of Unknown Significance Found during the 20-Year Activity of a Genetics Diagnostic Laboratory. Eur. J. Med. Genet. 2023, 66, 104847. [Google Scholar] [CrossRef]
| Gene | BC Risk * | OC Risk * | Other Cancer Risk |
|---|---|---|---|
| BRCA1 | 60–66% | 41–58% | Pancreatic cancer |
| BRCA2 | 55–61% | 15–16% | Pancreatic and Prostate cancer |
| ATM | 20–40% | 2–3% | Pancreatic, Prostate cancer |
| BARD1 | 20–40% | Not Assessed | Insufficient Evidence |
| BRIP1 | Not Assessed | 5–15% | Insufficient Evidence |
| CDH1 | 41–60% | Not Assessed | Hereditary diffuse gastric cancer |
| CHEK2 | 20–40% | Not Assessed | Colorectal, kidney, thyroid cancer |
| PALB2 | 41–60% | 3–5% | Pancreatic cancer |
| PTEN | 40–60% | Not Assessed | Colorectal, renal, thyroid cancer |
| RAD51C | 20–40% | 10–15% | Insufficient Evidence |
| RAD51D | 20–40% | 10–20% | Insufficient Evidence |
| TP53 | 60% | Not Assessed | Brain tumors, sarcoma, acute leukemia, adrenocortical tumors |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barili, V.; Ambrosini, E.; Bortesi, B.; Minari, R.; De Sensi, E.; Cannizzaro, I.R.; Taiani, A.; Michiara, M.; Sikokis, A.; Boggiani, D.; et al. Genetic Basis of Breast and Ovarian Cancer: Approaches and Lessons Learnt from Three Decades of Inherited Predisposition Testing. Genes 2024, 15, 219. https://doi.org/10.3390/genes15020219
Barili V, Ambrosini E, Bortesi B, Minari R, De Sensi E, Cannizzaro IR, Taiani A, Michiara M, Sikokis A, Boggiani D, et al. Genetic Basis of Breast and Ovarian Cancer: Approaches and Lessons Learnt from Three Decades of Inherited Predisposition Testing. Genes. 2024; 15(2):219. https://doi.org/10.3390/genes15020219
Chicago/Turabian StyleBarili, Valeria, Enrico Ambrosini, Beatrice Bortesi, Roberta Minari, Erika De Sensi, Ilenia Rita Cannizzaro, Antonietta Taiani, Maria Michiara, Angelica Sikokis, Daniela Boggiani, and et al. 2024. "Genetic Basis of Breast and Ovarian Cancer: Approaches and Lessons Learnt from Three Decades of Inherited Predisposition Testing" Genes 15, no. 2: 219. https://doi.org/10.3390/genes15020219