
Genome-Wide Detection for Runs of Homozygosity in Baoshan Pigs Using Whole Genome Resequencing

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
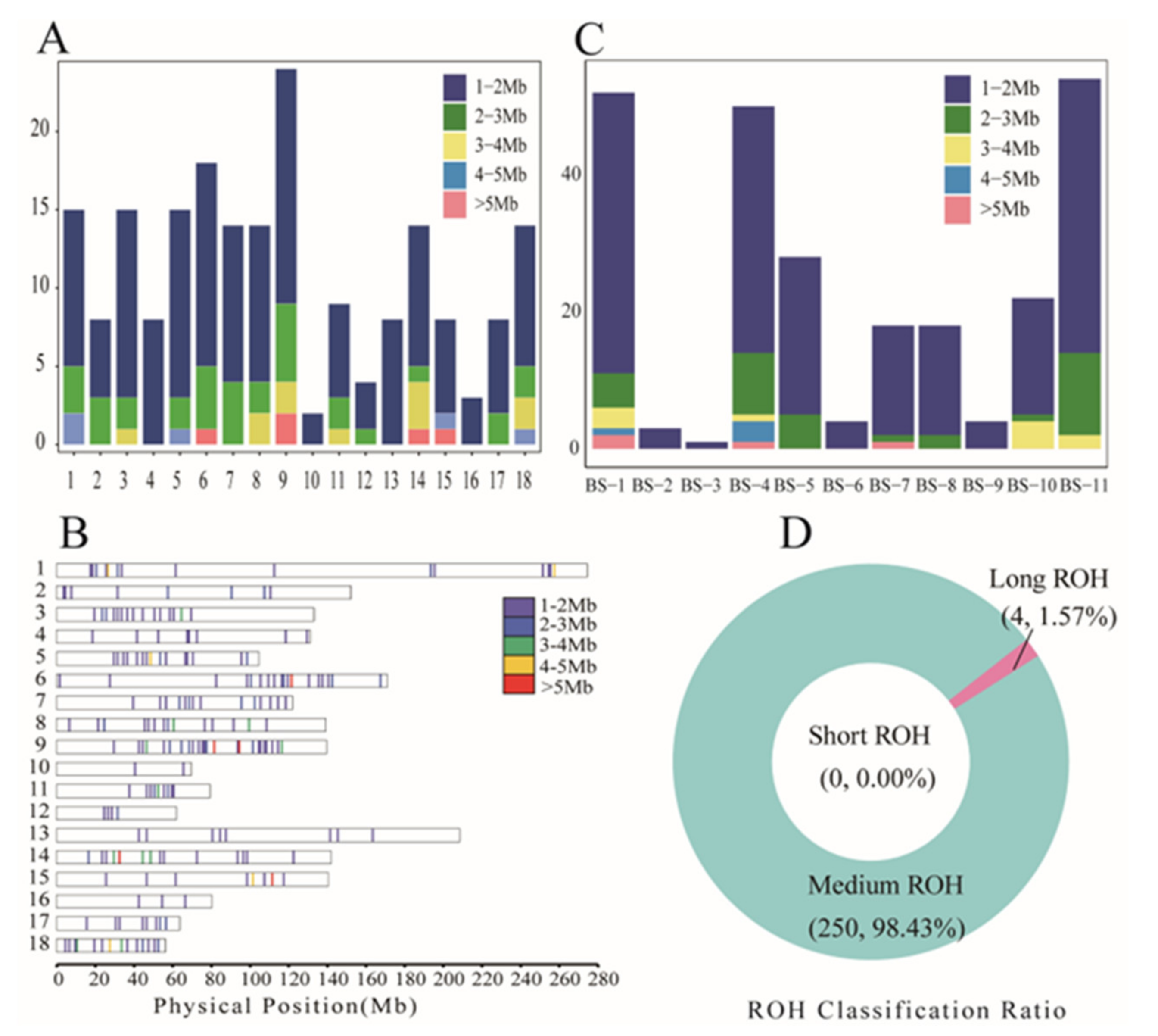
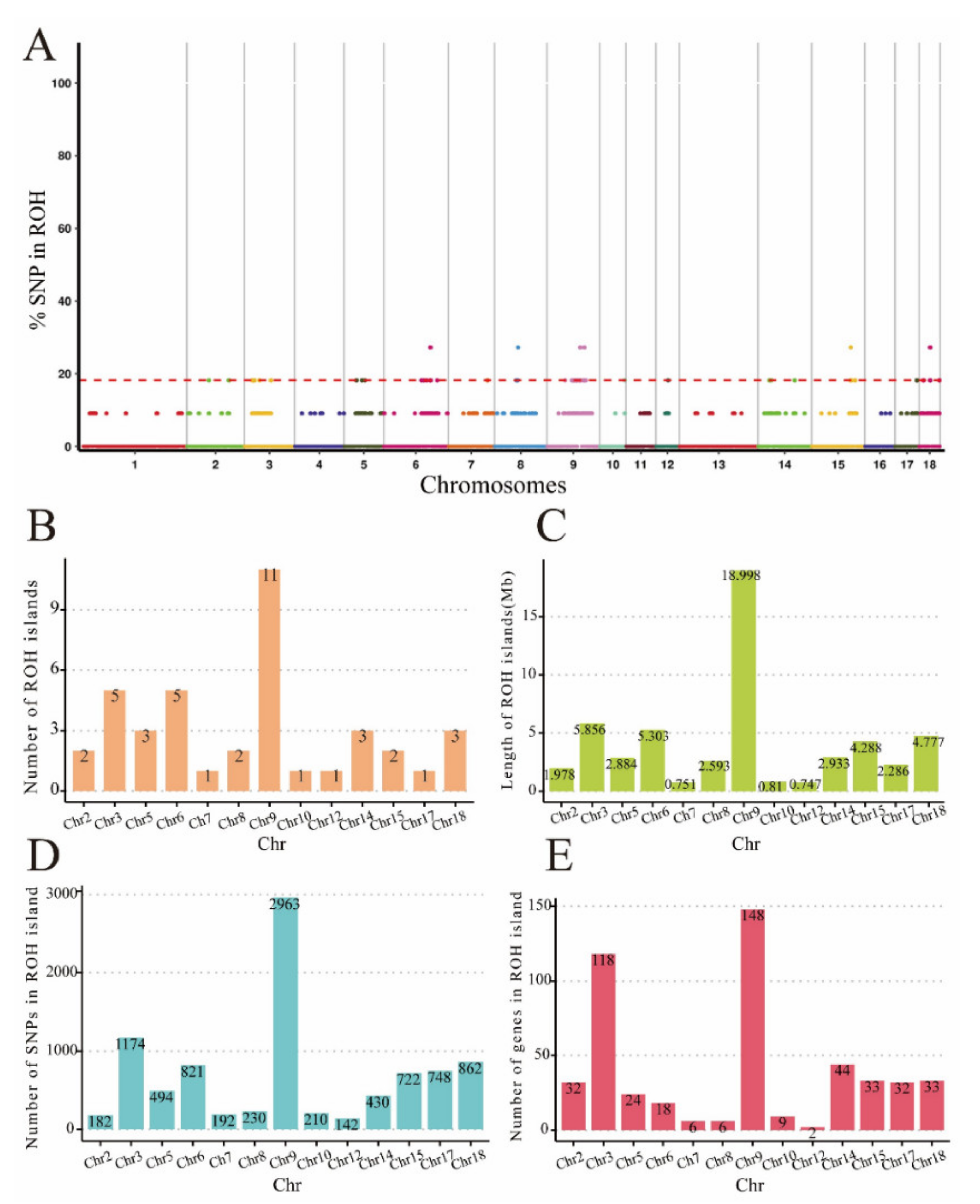
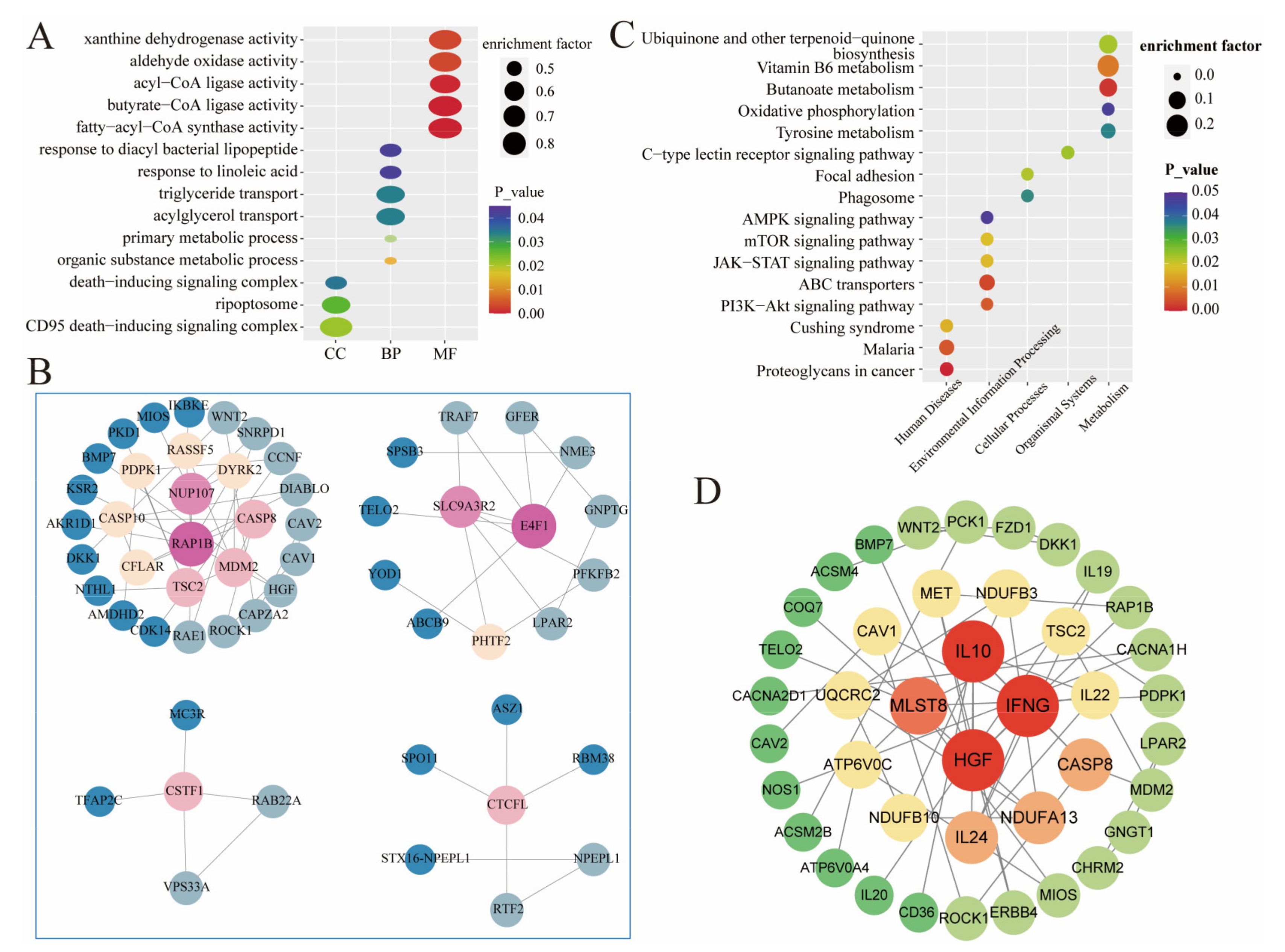
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, H.; Huang, T.; Zhang, Z.; Yang, B.; Jiang, C.; Wu, J.; Zhou, Z.; Zheng, H.; Xin, W.; Huang, M.; et al. Genome-wide association studies and meta-analysis reveal novel quantitative trait loci and pleiotropic loci for swine head-related traits. J. Anim. Sci. 2017, 95, 2354–2366. [Google Scholar] [CrossRef]
- Wu, X.; Zhou, R.; Zhang, W.; Cao, B.; Xia, J.; Wang, C.; Zhang, X.; Chu, M.; Yin, Z.; Ding, Y. Genome-wide scan for runs of homozygosity identifies candidate genes in Wannan Black pigs. Anim. Biosci. 2021, 34, 1895–1902. [Google Scholar] [CrossRef]
- Zhao, Q.B.; Lopez-Cortegano, E.; Oyelami, F.O.; Zhang, Z.; Ma, P.P.; Wang, Q.S.; Pan, Y.C. Conservation Priorities Analysis of Chinese Indigenous Pig Breeds in the Taihu Lake Region. Front. Genet. 2021, 12, 558873. [Google Scholar] [CrossRef]
- Garrido-Fernandez, A.; Leon-Camacho, M. Effect of season, feeding, and anatomical region on the triacylglycerol profile of Iberian pig fat. Food Chem. 2021, 361, 130070. [Google Scholar] [CrossRef]
- Pabst, R. The pig as a model for immunology research. Cell Tissue Res. 2020, 380, 287–304. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.; Wu, G.; Chen, T.; Huo, H.; Yuan, F.; Liu, L.; Ge, C.; Miao, Y. Genetic diversity of local Yunnan chicken breeds and their relationships with Red Junglefowl. Genet. Mol. Res. 2014, 13, 3371–3383. [Google Scholar] [CrossRef] [PubMed]
- Diao, S.; Huang, S.; Chen, Z.; Teng, J.; Ma, Y.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J.; Zhang, Z. Genome-Wide Signatures of Selection Detection in Three South China Indigenous Pigs. Genes 2019, 10, 346. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.T.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016, 10, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Bravo, I.; Wang, J. Inbreeding load and purging: Implications for the short-term survival and the conservation management of small populations. Heredity 2017, 118, 177–185. [Google Scholar] [CrossRef]
- Szmatola, T.; Jasielczuk, I.; Semik-Gurgul, E.; Szyndler-Nedza, M.; Blicharski, T.; Szulc, K.; Skrzypczak, E.; Gurgul, A. Detection of runs of homozygosity in conserved and commercial pig breeds in Poland. J. Anim. Breed. Genet. 2020, 137, 571–580. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Mtileni, B.; Dzama, K.; Nephawe, K.; Rhode, C. Estimates of effective population size and inbreeding in South African indigenous chicken populations: Implications for the conservation of unique genetic resources. Trop. Anim. Health Prod. 2016, 48, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Nosrati, M.; Asadollahpour Nanaei, H.; Javanmard, A.; Esmailizadeh, A. The pattern of runs of homozygosity and genomic inbreeding in world-wide sheep populations. Genomics 2021, 113, 1407–1415. [Google Scholar] [CrossRef]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef]
- Peripolli, E.; Munari, D.P.; Silva, M.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- Signer-Hasler, H.; Burren, A.; Ammann, P.; Drögemüller, C.; Flury, C. Runs of homozygosity and signatures of selection: A comparison among eight local Swiss sheep breeds. Anim. Genet. 2019, 50, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Szmatola, T.; Gurgul, A.; Jasielczuk, I.; Zabek, T.; Ropka-Molik, K.; Litwinczuk, Z.; Bugno-Poniewierska, M. A Comprehensive Analysis of Runs of Homozygosity of Eleven Cattle Breeds Representing Different Production Types. Animals 2019, 9, 1024. [Google Scholar] [CrossRef]
- Cesarani, A.; Gaspa, G.; Pauciullo, A.; Degano, L.; Vicario, D.; Macciotta, N.P.P. Genome-wide analysis of homozygosity regions in european simmental bulls. J. Anim. Breed. Genet. 2021, 138, 69–79. [Google Scholar] [CrossRef]
- Polak, G.; Gurgul, A.; Jasielczuk, I.; Szmatola, T.; Krupinski, J.; Bugno-Poniewierska, M. Suitability of Pedigree Information and Genomic Methods for Analyzing Inbreeding of Polish Cold-Blooded Horses Covered by Conservation Programs. Genes 2021, 12, 429. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, F.; Cardoso, T.F.; Marras, G.; Nicolazzi, E.L.; Rothschild, M.F.; Amills, M.; Consortium, A. Genome-wide patterns of homozygosity provide clues about the population history and adaptation of goats. Genet. Sel. Evol. 2018, 50, 59. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhou, R.; Wang, Y.; Zhang, W.; Zheng, X.; Zhao, G.; Zhang, X.; Yin, Z.; Ding, Y. Genome-wide scan for runs of homozygosity in Asian wild boars and Anqing six-end-white pigs. Anim. Genet. 2022, 53, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Howrigan, D.P.; Simonson, M.A.; Keller, M.C. Detecting autozygosity through runs of homozygosity: A comparison of three autozygosity detection algorithms. BMC Genom. 2011, 12, 460. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, G.; Yang, L.; Zhu, B.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Liu, G.E.; Li, J. Genomic Patterns of Homozygosity in Chinese Local Cattle. Sci. Rep. 2019, 9, 16977. [Google Scholar] [CrossRef]
- Dixit, S.P.; Singh, S.; Ganguly, I.; Bhatia, A.K.; Sharma, A.; Kumar, N.A.; Dang, A.K.; Jayakumar, S. Genome-Wide Runs of Homozygosity Revealed Selection Signatures in Bos indicus. Front. Genet. 2020, 11, 92. [Google Scholar] [CrossRef]
- Zhang, P.; Qiu, X.; Wang, L.; Zhao, F. Progress in Genomic Mating in Domestic Animals. Animals 2022, 12, 2306. [Google Scholar] [CrossRef]
- Severson, A.L.; Carmi, S.; Rosenberg, N.A. The Effect of Consanguinity on Between-Individual Identity-by-Descent Sharing. Genetics 2019, 212, 305–316. [Google Scholar] [CrossRef]
- Liu, G.; Sun, F.Z.; Zhu, F.X.; Feng, H.Y.; Han, X. Runs of homozygosity and its application on livestock genome study. Yi Chuan 2019, 41, 304–317. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Gomez-Raya, L.; Rodriguez, C.; Barragan, C.; Silio, L. Genomic inbreeding coefficients based on the distribution of the length of runs of homozygosity in a closed line of Iberian pigs. Genet. Sel. Evol. 2015, 47, 81. [Google Scholar] [CrossRef]
- Groenen, M.A.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.-J. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef]
- Herrero-Medrano, J.M.; Megens, H.J.; Groenen, M.A.; Ramis, G.; Bosse, M.; Perez-Enciso, M.; Crooijmans, R.P. Conservation genomic analysis of domestic and wild pig populations from the Iberian Peninsula. BMC Genet. 2013, 14, 106. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.; Souto, L.; Soares, A.M.; Fonseca, C. Genetic structure of the wild boar population in Portugal: Evidence of a recent bottleneck. Mamm. Biol. 2009, 74, 274–285. [Google Scholar] [CrossRef]
- Wu, F.; Sun, H.; Lu, S.; Gou, X.; Yan, D.; Xu, Z.; Zhang, Z.; Qadri, Q.R.; Zhang, Z.; Wang, Z. Genetic diversity and selection signatures within Diannan small-ear pigs revealed by next-generation sequencing. Front. Genet. 2020, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Shi, L.; Liu, J.; Deng, T.; Wang, L.; Liu, Y.; Zhao, F. Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes in Three Pig Breeds. Animals 2019, 9, 518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Q.; Xiao, Q.; Sun, H.; Gao, H.; Yang, Y.; Chen, J.; Li, Z.; Xue, M.; Ma, P.; et al. Distribution of runs of homozygosity in Chinese and Western pig breeds evaluated by reduced-representation sequencing data. Anim. Genet. 2018, 49, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Silio, L.; Rodriguez, M.C.; Fernandez, A.; Barragan, C.; Benitez, R.; Ovilo, C.; Fernandez, A.I. Measuring inbreeding and inbreeding depression on pig growth from pedigree or SNP-derived metrics. J. Anim. Breed. Genet. 2013, 130, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Saura, M.; Fernandez, A.; Varona, L.; Fernandez, A.I.; de Cara, M.A.; Barragan, C.; Villanueva, B. Detecting inbreeding depression for reproductive traits in Iberian pigs using genome-wide data. Genet. Sel. Evol. 2015, 47, 1. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wang, L.; Liu, J.; Deng, T.; Yan, H.; Zhang, L.; Liu, X.; Gao, H.; Hou, X.; Wang, L.; et al. Estimation of inbreeding and identification of regions under heavy selection based on runs of homozygosity in a Large White pig population. J. Anim. Sci. Biotechnol. 2020, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Valluzzi, C.; Rando, A.; Macciotta, N.P.P.; Gaspa, G.; Di Gregorio, P. The Nero Lucano Pig Breed: Recovery and Variability. Animals 2021, 11, 1331. [Google Scholar] [CrossRef] [PubMed]
- Zorc, M.; Skorput, D.; Gvozdanovic, K.; Margeta, P.; Karolyi, D.; Lukovic, Z.; Salajpal, K.; Savic, R.; Munoz, M.; Bovo, S.; et al. Genetic diversity and population structure of six autochthonous pig breeds from Croatia, Serbia, and Slovenia. Genet. Sel. Evol. 2022, 54, 30. [Google Scholar] [CrossRef] [PubMed]
- Forutan, M.; Ansari Mahyari, S.; Baes, C.; Melzer, N.; Schenkel, F.S.; Sargolzaei, M. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genom. 2018, 19, 98. [Google Scholar] [CrossRef]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Zhang, S.; Zhang, K.; Peng, X.; Xie, S.; Li, X.; Zhao, S.; Ma, Y. Genome-wide patterns of homozygosity and relevant characterizations on the population structure in Piétrain pigs. Genes 2020, 11, 577. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Bertolini, F.; Tinarelli, S.; Dall’Olio, S.; Nanni Costa, L.; Gallo, M.; Fontanesi, L. Comparative evaluation of genomic inbreeding parameters in seven commercial and autochthonous pig breeds. Animal 2020, 14, 910–920. [Google Scholar] [CrossRef]
- Krupa, E.; Moravcikova, N.; Krupova, Z.; Zakova, E. Assessment of the Genetic Diversity of a Local Pig Breed Using Pedigree and SNP Data. Genes 2021, 12, 1972. [Google Scholar] [CrossRef]
- Nothnagel, M.; Lu, T.T.; Kayser, M.; Krawczak, M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Hum. Mol. Genet. 2010, 19, 2927–2935. [Google Scholar] [CrossRef]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef]
- Hogler, W.; Martin, D.D.; Crabtree, N.; Nightingale, P.; Tomlinson, J.; Metherell, L.; Rosenfeld, R.; Hwa, V.; Rose, S.; Walker, J.; et al. IGFALS gene dosage effects on serum IGF-I and glucose metabolism, body composition, bone growth in length and width, and the pharmacokinetics of recombinant human IGF-I administration. J. Clin. Endocrinol. Metab. 2014, 99, E703-712. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, F.; Moradian, F.; Farhadi, A. Association of allelic polymorphisms of IGFALS gene with growth traits in Makouei and Ghezel sheep breeds. Trop. Anim. Health Prod. 2020, 52, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ren, J.; Huang, L. Characterization of the porcine insulin-like growth factor-binding protein, acid-labile subunit gene: Full-length cDNA and DNA sequence, polymorphisms and expression profile. J. Anim. Breed. Genet. 2007, 124, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Gozalo-Marcilla, M.; Buntjer, J.; Johnsson, M.; Batista, L.; Diez, F.; Werner, C.R.; Chen, C.Y.; Gorjanc, G.; Mellanby, R.J.; Hickey, J.M.; et al. Genetic architecture and major genes for backfat thickness in pig lines of diverse genetic backgrounds. Genet. Sel. Evol. 2021, 53, 76. [Google Scholar] [CrossRef] [PubMed]
- Xiang, G.; Huang, L.; Zhang, X.; Wang, N.; Wang, H.; Mu, Y.; Li, K.; Liu, Z. Molecular Characteristics and Promoter Analysis of Porcine COL1A1. Genes 2022, 13, 1971. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.R.; Tsai, S.; Hardison, N.; Motsinger-Reif, A.A.; Freking, B.A.; Nonneman, D.; Rohrer, G.; Piedrahita, J.A. Characterization of conserved and nonconserved imprinted genes in swine. Biol. Reprod. 2009, 81, 906–920. [Google Scholar] [CrossRef]
- Cheng, H.C.; Zhang, F.W.; Jiang, C.D.; Li, F.E.; Xiong, Y.Z.; Deng, C.Y. Isolation and imprinting analysis of the porcine DLX5 gene and its association with carcass traits. Anim. Genet. 2008, 39, 395–399. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Wang, F.; Hu, L.; Li, Y.; Wang, J.; Zhang, C.; Wang, S. Spatiotemporal expression of Wnt/β-catenin signaling during morphogenesis and odontogenesis of deciduous molar in miniature pig. Int. J. Biol. Sci. 2017, 13, 1082. [Google Scholar] [CrossRef]
- Fang, J.; Zhang, D.; Cao, J.W.; Zhang, L.; Liu, C.X.; Xing, Y.P.; Wang, F.; Xu, H.Y.; Wang, S.C.; Ling, Y.; et al. Pathways involved in pony body size development. BMC Genom. 2021, 22, 58. [Google Scholar] [CrossRef]
- Fan, Z.C.; Sartin, J.L.; Tao, Y.X. Molecular cloning and pharmacological characterization of porcine melanocortin-3 receptor. J. Endocrinol. 2008, 196, 139–148. [Google Scholar] [CrossRef]
- Edea, Z.; Kim, K.-S. A whole genomic scan to detect selection signatures between Berkshire and Korean native pig breeds. J. Anim. Sci. Technol. 2014, 56, 23. [Google Scholar] [CrossRef]
- Wang, D.; Liu, C.D.; Li, H.F.; Tian, M.L.; Pan, J.Q.; Shu, G.; Jiang, Q.Y.; Yin, Y.L.; Zhang, L. LSD1 mediates microbial metabolite butyrate-induced thermogenesis in brown and white adipose tissue. Metabolism 2020, 102, 154011. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Hao, X.; Xu, Z.; Sun, H.; Zhao, Q.; Cao, R.; Zhang, Z.; Ma, P.; Sun, Y.; Qi, Z.; et al. Genome-Wide Detection of Runs of Homozygosity in Laiwu Pigs Revealed by Sequencing Data. Front. Genet. 2021, 12, 629966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, L.; Zhou, M.; Su, S.; Dong, L.; Meng, X.; Li, X.; Wang, C. Assessing Population Structure and Signatures of Selection in Wanbei Pigs Using Whole Genome Resequencing Data. Animals 2022, 13, 13. [Google Scholar] [CrossRef]
- Zhang, R.; Feng, X.; Zhan, M.; Huang, C.; Chen, K.; Tang, X.; Kang, T.; Xiong, Y.; Lei, M. Transcription Factor Sp1 Promotes the Expression of Porcine ROCK1 Gene. Int. J. Mol. Sci. 2016, 17, 112. [Google Scholar] [CrossRef]
- Li, J.H.; Chu, X.H.; Guo, X.L.; Xu, N.Y. Radiation hybrid mapping of skeletal muscle calcium channel genes CACNB1 and CACNG1 to porcine chromosome 12 and CACNA2D1 to porcine chromosome 9. Anim. Genet. 2005, 36, 358–359. [Google Scholar] [CrossRef]
- Shimomura, Y.; Harada, M.; Goto, M.; Sugo, T.; Matsumoto, Y.; Abe, M.; Watanabe, T.; Asami, T.; Kitada, C.; Mori, M. Identification of neuropeptide W as the endogenous ligand for orphan G-protein-coupled receptors GPR7 and GPR8. J. Biol. Chem. 2002, 277, 35826–35832. [Google Scholar] [CrossRef]
- Dardente, H.; Wyse, C.A.; Birnie, M.J.; Dupre, S.M.; Loudon, A.S.; Lincoln, G.A.; Hazlerigg, D.G. A molecular switch for photoperiod responsiveness in mammals. Curr. Biol. 2010, 20, 2193–2198. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-L.; Li, Z.; Lin, T.-Y.; Wang, S.-W.; Wu, F.-J.; Luo, C.-W. Thyrostimulin-TSHR signaling promotes the proliferation of NIH: OVCAR-3 ovarian cancer cells via trans-regulation of the EGFR pathway. Sci. Rep. 2016, 6, 27471. [Google Scholar] [CrossRef]
- Du, X.; Yin, H.; Pan, Z.; Wu, W.; Shang, P.; Chamba, Y.; Li, Q. BMP7 is a candidate gene for reproductive traits in Yorkshire sows. Anim. Reprod. Sci. 2020, 221, 106598. [Google Scholar] [CrossRef]
- Yin, H.; Du, X.; Li, Q.; Pan, Z.; Wu, W.; Liu, H.; Li, Q. Variants in BMP7 and BMP15 3′-UTR s associated with reproductive traits in a large white pig population. Animals 2019, 9, 905. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Xie, S.Y.; Zhou, J.S.; Sun, G.R.; Lu, P.; Li, M. Polymorphisms of the bone morphogenetic protein 7 gene (BMP7) and association analysis with sow productive traits. Anim. Reprod. Sci. 2013, 142, 56–62. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Wu, X.; Xiang, D.; Zhang, W.; Wu, L.; Meng, X.; Huo, J.; Yin, Z.; Fu, G.; Zhao, G. Genome-Wide Detection for Runs of Homozygosity in Baoshan Pigs Using Whole Genome Resequencing. Genes 2024, 15, 233. https://doi.org/10.3390/genes15020233
Li W, Wu X, Xiang D, Zhang W, Wu L, Meng X, Huo J, Yin Z, Fu G, Zhao G. Genome-Wide Detection for Runs of Homozygosity in Baoshan Pigs Using Whole Genome Resequencing. Genes. 2024; 15(2):233. https://doi.org/10.3390/genes15020233
Chicago/Turabian StyleLi, Wenjun, Xudong Wu, Decai Xiang, Wei Zhang, Lingxiang Wu, Xintong Meng, Jinlong Huo, Zongjun Yin, Guowen Fu, and Guiying Zhao. 2024. "Genome-Wide Detection for Runs of Homozygosity in Baoshan Pigs Using Whole Genome Resequencing" Genes 15, no. 2: 233. https://doi.org/10.3390/genes15020233
APA StyleLi, W., Wu, X., Xiang, D., Zhang, W., Wu, L., Meng, X., Huo, J., Yin, Z., Fu, G., & Zhao, G. (2024). Genome-Wide Detection for Runs of Homozygosity in Baoshan Pigs Using Whole Genome Resequencing. Genes, 15(2), 233. https://doi.org/10.3390/genes15020233