
Comparative Analysis of Chloroplast Genomes of “Tiantai Wu-Yao” (Lindera aggregata) and Taxa of the Same Genus and Different Genera

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials & Methods
2.1. Sample Collection, DNA Extraction and Sequencing
2.2. Assembly and Annotation of the Chloroplast Genomes
2.3. Characteristics Analysis of Chloroplast Genomes
2.4. Comparative Genomics Analysis of the Chloroplast Genome
2.5. Phylogenetic Analysis
2.6. Divergence Time Estimated
3. Results
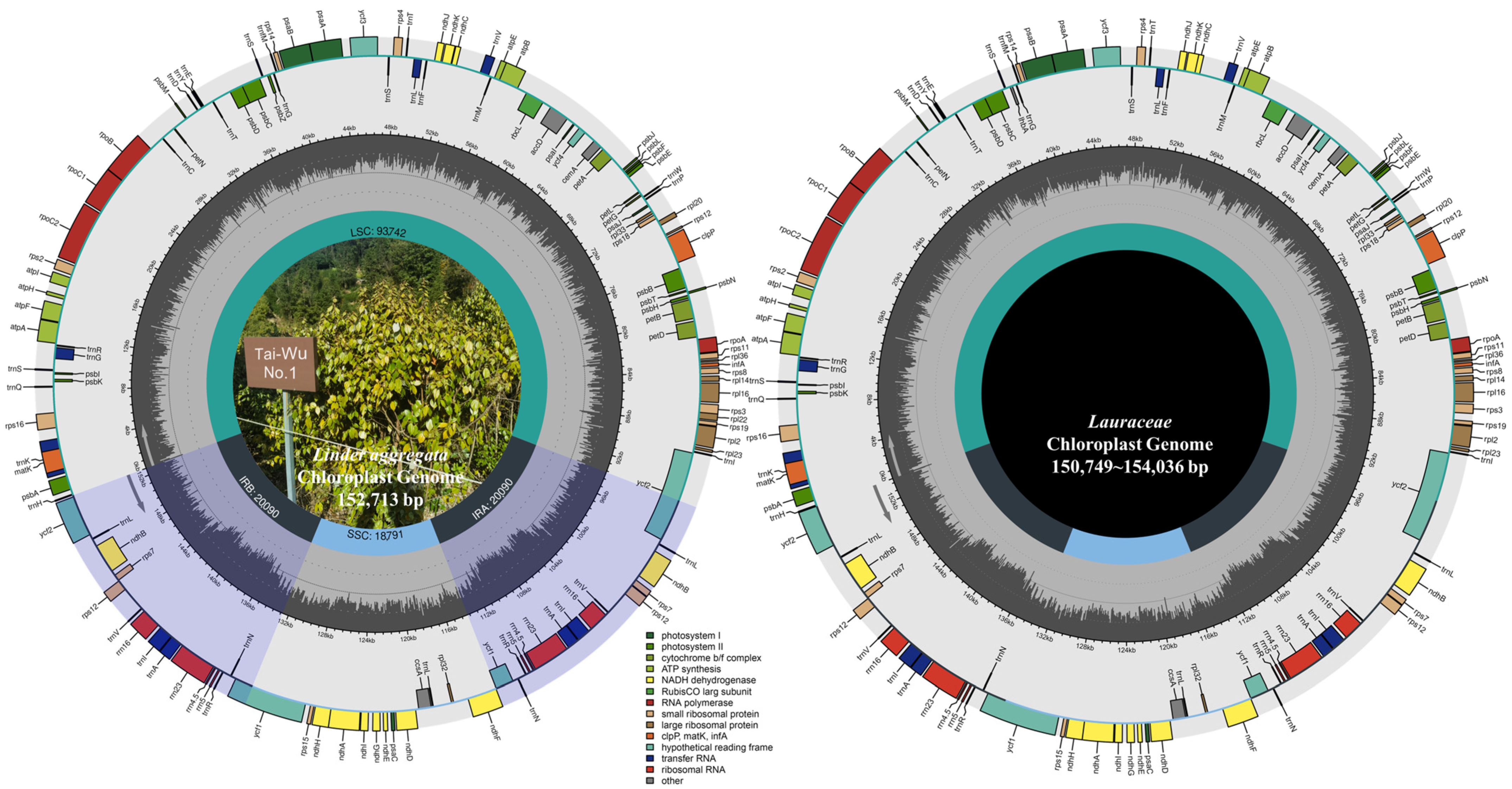
3.1. Analysis of Characteristics of Chloroplast Genomes
3.2. Expansion/Contraction Analysis of the Boundary
3.3. Comparative Chloroplast Genome Sequences Differences
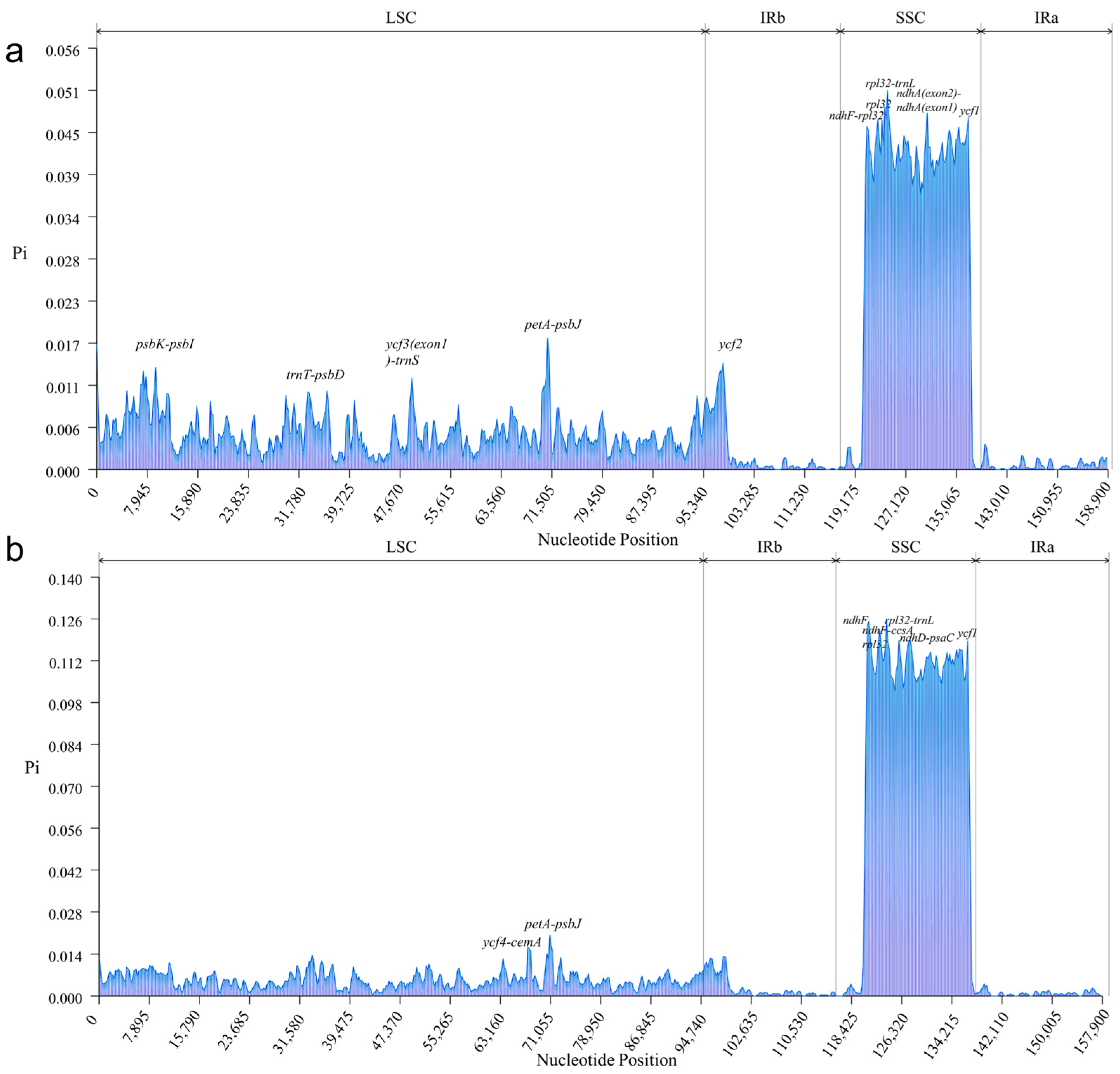
3.4. Analysis of a Highly Variable Region of Chloroplast Genomes
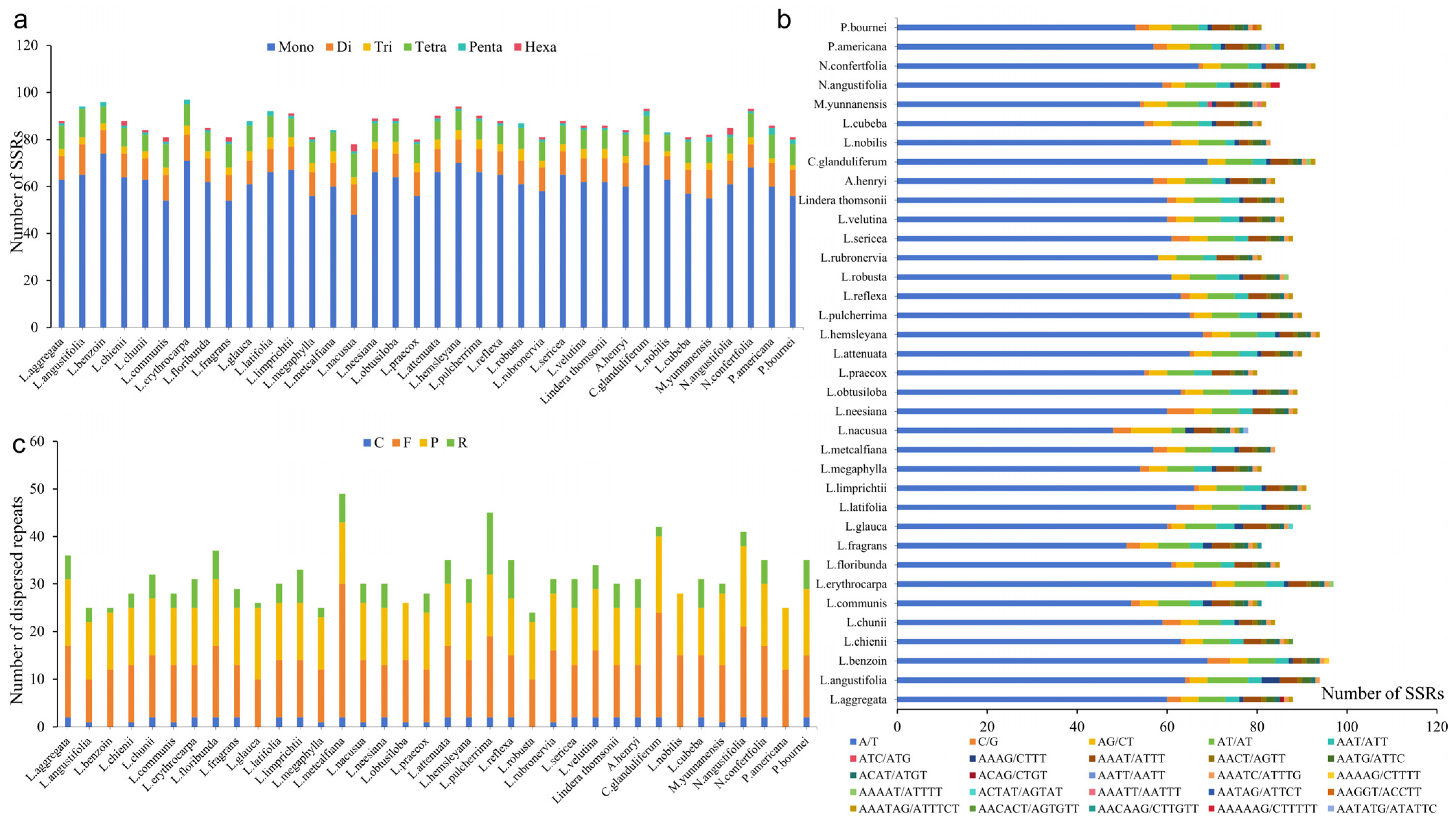
3.5. SSRs and Interspersed Repeat Sequence Analysis
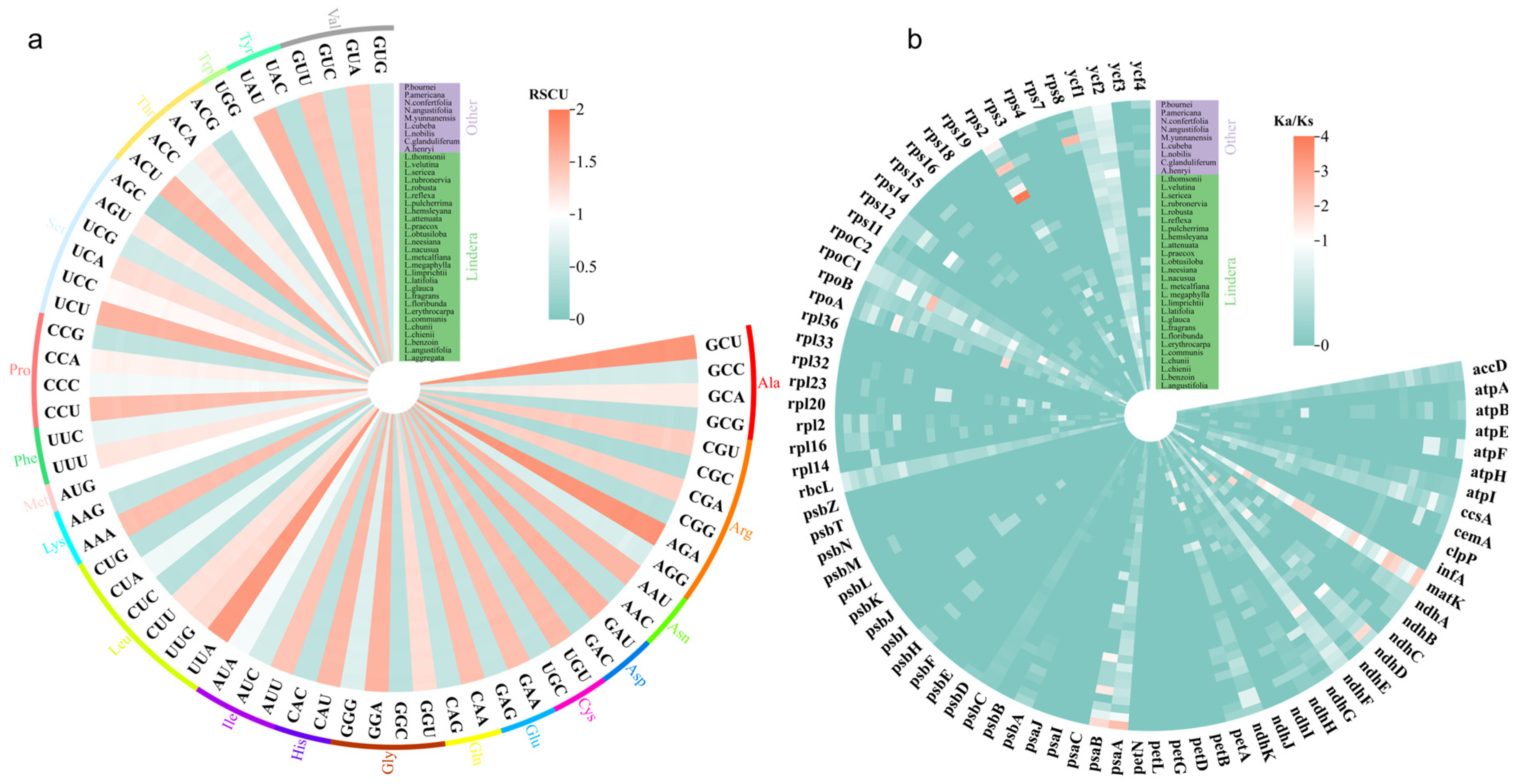
3.6. Analysis of Codon Bias and Selection Pressure in Chloroplast Genomes
3.7. Phylogenetic Trees and Divergence Time Analysis of the Chloroplast Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chanderbali, A.S.; van der Werff, H.; Renner, S.S. Phylogeny and Historical Biogeography of Lauraceae: Evidence from the Chloroplast and Nuclear Genomes. Ann. Mo. Bot. Gard. 2001, 88, 104–134. [Google Scholar] [CrossRef]
- van der Werff, H.; Richter, H.G. Toward an Improved Classification of Lauraceae. Ann. Mo. Bot. Gard. 1996, 83, 409–418. [Google Scholar] [CrossRef]
- Hung-Pin, T. A Study on the System of Lindera. J. Syst. Evol. 1987, 25, 161–171. [Google Scholar]
- Tao, Y.; Deng, Y.; Wang, P. Traditional uses, phytochemistry, pharmacology, processing methods and quality control of Lindera aggregata (Sims) Kosterm: A critical review. J. Ethnopharmacol. 2024, 318, 116954. [Google Scholar] [CrossRef]
- Lv, Y.; Zou, Y.; Zhang, X.; Liu, B.; Peng, X.; Chu, C. A review on the chemical constituents and pharmacological efficacies of Lindera aggregata (Sims) Kosterm. Front. Nutr. 2023, 9, 1071276. [Google Scholar] [CrossRef]
- Wang, C.; Dai, Y.; Yang, J.; Chou, G.; Wang, C.; Wang, Z. Treatment with total alkaloids from Radix Linderae reduces inflammation and joint destruction in type II collagen-induced model for rheumatoid arthritis. J. Ethnopharmacol. 2007, 111, 322–328. [Google Scholar] [CrossRef]
- Gan, L.-S.; Yao, W.; Mo, J.-X.; Zhou, C.-X. Alkaloids from Lindera aggregata. Nat. Prod. Commun. 2009, 4, 1934578X0900400111. [Google Scholar] [CrossRef]
- Han, H.; Xu, B.; Amin, A.; Li, H.; Yu, X.; Gong, M.; Zhang, L. Quercetin-3-O-α-L-rhamnopyranoside derived from the leaves of Lindera aggregata (Sims) Kosterm. evokes the autophagy-induced nuclear factor erythroid 2-related factor 2 antioxidant pathway in human umbilical vein endothelial cells. Int. J. Mol. Med. 2019, 43, 461–474. [Google Scholar] [CrossRef]
- Wu, Y.; Zheng, Y.; Liu, X.; Han, Z.; Ren, Y.; Gan, L.; Zhou, C.; Luan, L. Separation and quantitative determination of sesquiterpene lactones in Lindera aggregata (Wu-yao) by ultra-performance LC-MS/MS. J. Sep. Sci. 2010, 33, 1072–1078. [Google Scholar] [CrossRef]
- Xu, C.; Yang, B.; Zhu, W.; Li, X.; Tian, J.; Zhang, L. Characterisation of polyphenol constituents of Linderae aggregata leaves using HPLC fingerprint analysis and their antioxidant activities. Food Chem. 2015, 186, 83–89. [Google Scholar] [CrossRef]
- Duan, C.; Guo, J.-M.; Dai, Y.; Xia, Y.-F. The absorption enhancement of norisoboldine in the duodenum of adjuvant-induced arthritis rats involves the impairment of P-glycoprotein. Biopharm. Drug Dispos. 2017, 38, 75–83. [Google Scholar] [CrossRef]
- Weng, M.; You, S.; Luo, J.; Lin, Z.; Chen, T.; Peng, X.; Qiu, B. Antibacterial mechanism of polysaccharides from the leaves of Lindera aggregata (Sims) Kosterm. by metabolomics based on HPLC/MS. Int. J. Biol. Macromol. 2022, 221, 303–313. [Google Scholar] [CrossRef]
- Salleh, W.; Hakimi, W.M. Lindera aggregata (Sims) Kosterm: Review on phytochemistry and biological activities. Bol. Latinoam. Y Del Caribe De Plantas Med. Y Aromat. 2020, 19, 527–541. [Google Scholar] [CrossRef]
- Wei, Z.-F.; Tong, B.; Xia, Y.-F.; Lu, Q.; Chou, G.-X.; Wang, Z.-T.; Dai, Y. Norisoboldine Suppresses Osteoclast Differentiation through Preventing the Accumulation of TRAF6-TAK1 Complexes and Activation of MAPKs/NF-κB/c-Fos/NFATc1 Pathways. PLoS ONE 2013, 8, e59171. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Cai, X.; Chen, Q.; Zhou, H.; Cai, Y.; Ben, A. Factors Affecting Synonymous Codon Usage Bias in Chloroplast Genome of Oncidium Gower Ramsey. Evol. Bioinform. 2011, 7, EBO.S8092. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Bock, R.; Knoop, V. Genomics of Chloroplasts and Mitochondria; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; Volume 35. [Google Scholar]
- Song, Y.; Yu, W.B.; Tan, Y.H.; Jin, J.J.; Wang, B.; Yang, J.B.; Liu, B.; Corlett, R.T. Plastid phylogenomics improve phylogenetic resolution in the Lauraceae. J. Syst. Evol. 2020, 58, 423–439. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, Y.; Tng, D.Y.; Zhou, J.; Zhang, Y.; Wang, Z.; Li, P.; Wang, Z. Comparative chloroplast genomics of Litsea Lam. (Lauraceae) and its phylogenetic implications. Forests 2021, 12, 744. [Google Scholar] [CrossRef]
- Tian, X.; Ye, J.; Song, Y. Plastome sequences help to improve the systematic position of trinerved Lindera species in the family Lauraceae. PeerJ 2019, 7, e7662. [Google Scholar] [CrossRef]
- Hinsinger, D.D.; Strijk, J.S. Toward phylogenomics of Lauraceae: The complete chloroplast genome sequence of Litsea glutinosa (Lauraceae), an invasive tree species on Indian and Pacific Ocean islands. Plant Gene 2017, 9, 71–79. [Google Scholar] [CrossRef]
- Zhao, M.-L.; Song, Y.; Ni, J.; Yao, X.; Tan, Y.-H.; Xu, Z.-F. Comparative chloroplast genomics and phylogenetics of nine Lindera species (Lauraceae). Sci. Rep. 2018, 8, 8844. [Google Scholar] [CrossRef]
- Song, Y.; Dong, W.; Liu, B.; Xu, C.; Yao, X.; Gao, J.; Corlett, R.T. Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machilus balansae in the family Lauraceae. Front. Plant Sci. 2015, 6, 662. [Google Scholar] [CrossRef]
- Song, Y.; Yao, X.; Liu, B.; Tan, Y.; Corlett, R.T. Complete plastid genome sequences of three tropical Alseodaphne trees in the family Lauraceae. Holzforschung 2018, 72, 337–345. [Google Scholar] [CrossRef]
- Song, Y.; Yao, X.; Tan, Y.; Gan, Y.; Corlett, R.T. Complete chloroplast genome sequence of the avocado: Gene organization, comparative analysis, and phylogenetic relationships with other Lauraceae. Can. J. For. Res. 2016, 46, 1293–1301. [Google Scholar] [CrossRef]
- Song, Y.; Yu, W.-B.; Tan, Y.; Liu, B.; Yao, X.; Jin, J.; Padmanaba, M.; Yang, J.-B.; Corlett, R.T. Evolutionary comparisons of the chloroplast genome in Lauraceae and insights into loss events in the Magnoliids. Genome Biol. Evol. 2017, 9, 2354–2364. [Google Scholar] [CrossRef]
- Yuan, L.; Sun, Y.; Zhou, S.; Wang, F.; Sun, C. Characterization of the complete chloroplast genome of Lindera aggregata. Mitochondrial DNA Part B 2021, 6, 3055–3056. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Christophel, D.C.; Conran, J.G.; Li, H.-W. Phylogenetic relationships within the ‘core’ Laureae (Litsea complex, Lauraceae) inferred from sequences of the chloroplast gene matK and nuclear ribosomal DNA ITS regions. Plant Syst. Evol. 2004, 246, 19–34. [Google Scholar] [CrossRef]
- Li, J.; Christophel, D.C. Systematic relationships within the Litsea complex (Lauraceae): A cladistic analysis on the basis of morphological and leaf cuticle data. Aust. Syst. Bot. 2000, 13, 1–13. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; DePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Zheng, S.; Poczai, P.; Hyvönen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An online program for the versatile plotting of organelle genomes. Front. Genet. 2020, 11, 576124. [Google Scholar] [CrossRef]
- Liu, S.; Ni, Y.; Li, J.; Zhang, X.; Yang, H.; Chen, H.; Liu, C. CPGView: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 2023, 23, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32 (Suppl. S2), W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Sánchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization by One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, gkad359. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Xiong, B.; Zhang, L.; Xie, L.; Li, L.; He, X.; Niu, Y.; Zhang, T.; Liao, S.; Dong, S.; Zhang, Z. Genome of Lindera glauca provides insights into the evolution of biosynthesis genes for aromatic compounds. iScience 2022, 25, 104761. [Google Scholar] [CrossRef]
- Jiang, R.; Chen, X.; Liao, X.; Peng, D.; Han, X.; Zhu, C.; Wang, P.; Hufnagel, D.E.; Wang, L.; Li, K.; et al. A Chromosome-Level Genome of the Camphor Tree and the Underlying Genetic and Climatic Factors for Its Top-Geoherbalism. Front. Plant Sci. 2022, 13, 827890. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Tao, L.; Duan, H.; Tao, K.; Luo, Y.; Li, Q.; Li, L. Complete chloroplast genome structural characterization of two Phalaenopsis (Orchidaceae) species and comparative analysis with their alliance. BMC Genom. 2023, 24, 359. [Google Scholar] [CrossRef]
- Koch, M.A.; Haubold, B.; Mitchell-Olds, T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 2000, 17, 1483–1498. [Google Scholar] [CrossRef]
- Funk, H.T.; Berg, S.; Krupinska, K.; Maier, U.G.; Krause, K. Complete DNA sequences of the plastid genomes of two parasitic flowering plant species, Cuscuta reflexa and Cuscuta gronovii. BMC Plant Biol. 2007, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-J.; Cheng, C.-L.; Chang, C.-C.; Wu, C.-L.; Su, T.-M.; Chaw, S.-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Y.; Cho, S.-T.; Haryono, M.; Kuo, C.-H. Complete chloroplast genome sequence of common bermudagrass (Cynodon dactylon (L.) Pers.) and comparative analysis within the family Poaceae. PLoS ONE 2017, 12, e0179055. [Google Scholar]
- Ren, J.; Tian, J.; Jiang, H.; Zhu, X.-X.; Mutie, F.M.; Wanga, V.O.; Ding, S.-X.; Yang, J.-X.; Dong, X.; Chen, L.-L. Comparative and phylogenetic analysis based on the chloroplast genome of Coleanthus subtilis (Tratt.) Seidel, a protected rare species of monotypic genus. Front. Plant Sci. 2022, 13, 828467. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhu, M.; Su, Y.; Wang, T. A Large Intergenic Spacer Leads to the Increase in Genome Size and Sequential Gene Movement around IR/SC Boundaries in the Chloroplast Genome of Adiantum malesianum (Pteridaceae). Int. J. Mol. Sci. 2022, 23, 15616. [Google Scholar] [CrossRef]
- Cai, H.; Xu, R.; Tian, P.; Zhang, M.; Zhu, L.; Yin, T.; Zhang, H.; Liu, X. Complete Chloroplast Genomes and the Phylogenetic Analysis of Three Native Species of Paeoniaceae from the Sino-Himalayan Flora Subkingdom. Int. J. Mol. Sci. 2023, 25, 257. [Google Scholar] [CrossRef]
- Liu, X.; Bai, Y.; Wang, Y.; Chen, Y.; Dong, W.; Zhang, Z. Complete chloroplast genome of Hypericum perforatum and dynamic evolution in Hypericum (Hypericaceae). Int. J. Mol. Sci. 2023, 24, 16130. [Google Scholar] [CrossRef]
- Rohwer, J.G.; Li, J.; Rudolph, B.; Schmidt, S.A.; van der Werff, H.; Li, H.-W. Is Persea (Lauraceae) monophyletic? Evidence from nuclear ribosomal ITS sequences. Taxon 2009, 58, 1153–1167. [Google Scholar] [CrossRef]
- Mo, Y.-Q.; Li, L.; Li, J.-W.; Rohwer, J.G.; Li, H.-W.; Li, J. Alseodaphnopsis: A new genus of Lauraceae based on molecular and morphological evidence. PLoS ONE 2017, 12, e0186545. [Google Scholar] [CrossRef]
- Li, L.; Li, J.; Rohwer, J.G.; van der Werff, H.; Wang, Z.-H.; Li, H.-W. Molecular phylogenetic analysis of the Persea group (Lauraceae) and its biogeographic implications on the evolution of tropical and subtropical Amphi-Pacific disjunctions. Am. J. Bot. 2011, 98, 1520–1536. [Google Scholar] [CrossRef]
- Hu, J.; Gui, S.; Zhu, Z.; Wang, X.; Ke, W.; Ding, Y. Genome-wide identification of SSR and SNP markers based on whole-genome re-sequencing of a Thailand wild sacred lotus (Nelumbo nucifera). PLoS ONE 2015, 10, e0143765. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Panesar, P.S.; Bera, M.B.; Kaur, V. Simple sequence repeat markers in genetic divergence and marker-assisted selection of rice cultivars: A review. Crit. Rev. Food Sci. Nutr. 2015, 55, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, X.; Meng, H.; Li, J.; Zhang, D.; Liu, C. Comparative chloroplast genomes of Paris Sect. Marmorata: Insights into repeat regions and evolutionary implications. BMC Genom. 2018, 19, 133–144. [Google Scholar] [CrossRef]
- Park, I.; Choi, B.; Weiss-Schneeweiss, H.; So, S.; Myeong, H.-H.; Jang, T.-S. Comparative analyses of complete chloroplast genomes and karyotypes of allotetraploid Iris koreana and its putative diploid parental species (Iris series Chinenses, Iridaceae). Int. J. Mol. Sci. 2022, 23, 10929. [Google Scholar] [CrossRef]
- Boël, G.; Letso, R.; Neely, H.; Price, W.N.; Wong, K.-H.; Su, M.; Luff, J.D.; Valecha, M.; Everett, J.K.; Acton, T.B. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 2016, 529, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Zhang, Q.; Wang, C.; Li, F.; Tian, F.; Lu, Y.; Hu, Y.; Yang, H.; Cui, G. Analysis of codon usage patterns of the chloroplast genome in Delphinium grandiflorum L. reveals a preference for AT-ending codons as a result of major selection constraints. PeerJ 2021, 9, e10787. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chen, H.-H.; Tang, L.-Z.; Khine, P.K.; Han, L.-H.; Song, Y.; Tan, Y.-H. Plastid genome evolution of a monophyletic group in the subtribe Lauriineae (Laureae, Lauraceae). Plant Divers. 2022, 44, 377–388. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Chen, Z.; Jiang, J.; Li, X.; Zeng, W. Comparative Analysis of Chloroplast Genomes of “Tiantai Wu-Yao” (Lindera aggregata) and Taxa of the Same Genus and Different Genera. Genes 2024, 15, 263. https://doi.org/10.3390/genes15030263
Shi Y, Chen Z, Jiang J, Li X, Zeng W. Comparative Analysis of Chloroplast Genomes of “Tiantai Wu-Yao” (Lindera aggregata) and Taxa of the Same Genus and Different Genera. Genes. 2024; 15(3):263. https://doi.org/10.3390/genes15030263
Chicago/Turabian StyleShi, Yujie, Zhen Chen, Jingyong Jiang, Xiaobai Li, and Wei Zeng. 2024. "Comparative Analysis of Chloroplast Genomes of “Tiantai Wu-Yao” (Lindera aggregata) and Taxa of the Same Genus and Different Genera" Genes 15, no. 3: 263. https://doi.org/10.3390/genes15030263