
Drug Repurposing and Lysosomal Storage Disorders: A Trick to Treat

 , , and
, , and
Abstract
:1. Introduction
2. Drug Repositioning for Lysosomal Storage Disorders: Still a Niche Field
3. Shooting at Random: The Valuable Results of High-Throughput Screening
3.1. Repositioning HTS-Identified Drugs as Pharmacological Chaperones
3.2. Repositioning HTS-Identified Drugs as Regulators of Lysosomal Function
4. Meet Serendipity Halfway: Targeted Drug Repositioning
4.1. Symptoms as the Compass
4.2. Mining Transcriptomics and Pathways
4.3. Using Drugs as a Template: Exploring Similarities in the Chemical Space
5. Expanding Human Skills: In Silico Models
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Thomas, S.; Caplan, A. The Orphan Drug Act Revisited. JAMA 2019, 321, 833. [Google Scholar] [CrossRef] [PubMed]
- Hageman, I.C.; Van Rooij, I.A.L.M.; De Blaauw, I.; Trajanovska, M.; King, S.K. A Systematic Overview of Rare Disease Patient Registries: Challenges in Design, Quality Management, and Maintenance. Orphanet J. Rare Dis. 2023, 18, 106. [Google Scholar] [CrossRef]
- Kölker, S.; Gleich, F.; Mütze, U.; Opladen, T. Rare Disease Registries Are Key to Evidence-Based Personalized Medicine: Highlighting the European Experience. Front. Endocrinol. 2022, 13, 832063. [Google Scholar] [CrossRef] [PubMed]
- Slebodnik, M. Orphanet: The Portal for Rare Diseases and Orphan Drugs. Ref. Rev. 2009, 23, 45–46. [Google Scholar] [CrossRef]
- Rath, A.; Olry, A.; Dhombres, F.; Brandt, M.M.; Urbero, B.; Ayme, S. Representation of Rare Diseases in Health Information Systems: The Orphanet Approach to Serve a Wide Range of End Users. Hum. Mutat. 2012, 33, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.Org: Online Mendelian Inheritance in Man (OMIM®), an Online Catalog of Human Genes and Genetic Disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef]
- Haendel, M.; Vasilevsky, N.; Unni, D.; Bologa, C.; Harris, N.; Rehm, H.; Hamosh, A.; Baynam, G.; Groza, T.; McMurry, J.; et al. How Many Rare Diseases Are There? Nat. Rev. Drug Discov. 2020, 19, 77–78. [Google Scholar] [CrossRef]
- Rahit, K.M.T.H.; Tarailo-Graovac, M. Genetic Modifiers and Rare Mendelian Disease. Genes 2020, 11, 239. [Google Scholar] [CrossRef]
- Boycott, K.M.; Rath, A.; Chong, J.X.; Hartley, T.; Alkuraya, F.S.; Baynam, G.; Brookes, A.J.; Brudno, M.; Carracedo, A.; den Dunnen, J.T.; et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am. J. Hum. Genet. 2017, 100, 695–705. [Google Scholar] [CrossRef]
- Nestler-Parr, S.; Korchagina, D.; Toumi, M.; Pashos, C.L.; Blanchette, C.; Molsen, E.; Morel, T.; Simoens, S.; Kaló, Z.; Gatermann, R.; et al. Challenges in Research and Health Technology Assessment of Rare Disease Technologies: Report of the ISPOR Rare Disease Special Interest Group. Value Health 2018, 21, 493–500. [Google Scholar] [CrossRef]
- Monticelli, M.; Francisco, R.; Brasil, S.; Marques-da-Silva, D.; Rijoff, T.; Pascoal, C.; Jaeken, J.; Videira, P.A.; Dos Reis Ferreira, V. Stakeholders’ Views on Drug Development: The Congenital Disorders of Glycosylation Community Perspective. Orphanet J. Rare Dis. 2022, 17, 303. [Google Scholar] [CrossRef]
- Szegedi, M.; Zelei, T.; Arickx, F.; Bucsics, A.; Cohn-Zanchetta, E.; Fürst, J.; Kamusheva, M.; Kawalec, P.; Petrova, G.; Slaby, J.; et al. The European Challenges of Funding Orphan Medicinal Products. Orphanet J. Rare Dis. 2018, 13, 184. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.-W. Development of Orphan Drugs for Rare Disease. Clin. Exp. Pediatr. 2023. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug Repositioning: Identifying and Developing New Uses for Existing Drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug Repurposing from the Perspective of Pharmaceutical Companies. Br. J. Pharmacol. 2018, 175, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Nosengo, N. Can You Teach Old Drugs New Tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844. [Google Scholar] [CrossRef]
- Hay Mele, B.; Citro, V.; Andreotti, G.; Cubellis, M.V. Drug Repositioning Can Accelerate Discovery of Pharmacological Chaperones. Orphanet J. Rare Dis. 2015, 10, 55. [Google Scholar] [CrossRef]
- Ratner, L. Glucosidase Inhibitors for Treatment of HIV-1 Infection. AIDS Res. Hum. Retroviruses 1992, 8, 165–173. [Google Scholar] [CrossRef]
- Liguori, L.; Monticelli, M.; Allocca, M.; Mele, B.H.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef]
- Andreotti, G.; Guarracino, M.R.; Cammisa, M.; Correra, A.; Cubellis, M.V. Prediction of the Responsiveness to Pharmacological Chaperones: Lysosomal Human α-Galactosidase, a Case of Study. Orphanet J. Rare Dis. 2010, 5, 36. [Google Scholar] [CrossRef]
- Andreotti, G.; Citro, V.; De Crescenzo, A.; Orlando, P.; Cammisa, M.; Correra, A.; Cubellis, M.V. Therapy of Fabry Disease with Pharmacological Chaperones: From in Silico Predictions to in Vitro Tests. Orphanet J. Rare Dis. 2011, 6, 66. [Google Scholar] [CrossRef]
- Online Resource, Rare Diseases and Alignment with Terminologies and Databases. Available online: https://www.orphadata.com/alignments/ (accessed on 13 December 2023).
- Shimizu, T.; Schutt, C.R.; Izumi, Y.; Tomiyasu, N.; Omahdi, Z.; Kano, K.; Takamatsu, H.; Aoki, J.; Bamba, T.; Kumanogoh, A.; et al. Direct Activation of Microglia by β-Glucosylceramide Causes Phagocytosis of Neurons That Exacerbates Gaucher Disease. Immunity 2023, 56, 307–319.e8. [Google Scholar] [CrossRef] [PubMed]
- Taranta, A.; Elmonem, M.A.; Bellomo, F.; De Leo, E.; Boenzi, S.; Janssen, M.J.; Jamalpoor, A.; Cairoli, S.; Pastore, A.; De Stefanis, C.; et al. Benefits and Toxicity of Disulfiram in Preclinical Models of Nephropathic Cystinosis. Cells 2021, 10, 3294. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, Y.R.; Robinson, P.N.; Gahl, W.A.; Avillach, P.; Baynam, G.; Cederroth, H.; Goodwin, R.M.; Groft, S.C.; Hansson, M.G.; Harris, N.L.; et al. The Case for Open Science: Rare Diseases. JAMIA Open 2020, 3, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Van Eck, N.J.; Waltman, L. Software Survey: VOSviewer, a Computer Program for Bibliometric Mapping. Scientometrics 2010, 84, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Olarte-Avellaneda, S.; Cepeda Del Castillo, J.; Rojas-Rodriguez, A.F.; Sánchez, O.; Rodríguez-López, A.; Suárez García, D.A.; Pulido, L.M.S.; Alméciga-Díaz, C.J. Bromocriptine as a Novel Pharmacological Chaperone for Mucopolysaccharidosis IV A. ACS Med. Chem. Lett. 2020, 11, 1377–1385. [Google Scholar] [CrossRef]
- Michael Besser, G.; Pfeiffer, R.F.; Thorner, M.O. ANNIVERSARY REVIEW: 50 Years since the Discovery of Bromocriptine. Eur. J. Endocrinol. 2018, 179, R69–R75. [Google Scholar] [CrossRef]
- Higaki, K.; Ninomiya, H.; Suzuki, Y.; Nanba, E. Candidate Molecules for Chemical Chaperone Therapy of G M1 -Gangliosidosis. Future Med. Chem. 2013, 5, 1551–1558. [Google Scholar] [CrossRef]
- Laur, D.; Pichard, S.; Bekri, S.; Caillaud, C.; Froissart, R.; Levade, T.; Roubertie, A.; Desguerre, I.; Héron, B.; Auvin, S. Natural History of GM1 Gangliosidosis—Retrospective Cohort Study of 61 French Patients from 1998 to 2019. J. Inherit. Metab. Dis. 2023, 46, 972–981. [Google Scholar] [CrossRef]
- Miura, K.; Onodera, C.; Takagi, M.; Koyama, R.; Hirano, T.; Nishio, T.; Hakamata, W. Screening, Synthesis, and Evaluation of Novel Isoflavone Derivatives as Inhibitors of Human Golgi β-Galactosidase. Chem. Pharm. Bull. 2020, 68, 753–761. [Google Scholar] [CrossRef]
- Buratti, E.; Peruzzo, P.; Braga, L.; Zanin, I.; Stuani, C.; Goina, E.; Romano, M.; Giacca, M.; Dardis, A. Deferoxamine Mesylate Improves Splicing and GAA Activity of the Common c.-32-13T>G Allele in Late-Onset PD Patient Fibroblasts. Mol. Ther. Methods Clin. Dev. 2021, 20, 227–236. [Google Scholar] [CrossRef]
- Gort, L.; Coll, M.J.; Chabás, A. Glycogen Storage Disease Type II in Spanish Patients: High Frequency of c.1076-1G>C Mutation. Mol. Genet. Metab. 2007, 92, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Herzog, A.; Hartung, R.; Reuser, A.J.J.; Hermanns, P.; Runz, H.; Karabul, N.; Gökce, S.; Pohlenz, J.; Kampmann, C.; Lampe, C.; et al. A Cross-Sectional Single-Centre Study on the Spectrum of Pompe Disease, German Patients: Molecular Analysis of the GAA Gene, Manifestation and Genotype-Phenotype Correlations. Orphanet J. Rare Dis. 2012, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.R.; Gläser, D.; Schmidt, S.; Vorgerd, M.; Winterholler, M.; Eger, K.; Zierz, S.; Deschauer, M. Molecular Diagnosis of German Patients with Late-onset Glycogen Storage Disease Type II. J. Inherit. Metab. Dis. 2008, 31, 261–265. [Google Scholar] [CrossRef]
- Montalvo, A.L.E.; Bembi, B.; Donnarumma, M.; Filocamo, M.; Parenti, G.; Rossi, M.; Merlini, L.; Buratti, E.; De Filippi, P.; Dardis, A.; et al. Mutation Profile of theGAA Gene in 40 Italian Patients with Late Onset Glycogen Storage Disease Type II. Hum. Mutat. 2006, 27, 999–1006. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Fanin, M.; Tasca, E.; Angelini, C. Molecular Pathology and Enzyme Processing in Various Phenotypes of Acid Maltase Deficiency. Neurology 2008, 70, 617–626. [Google Scholar] [CrossRef]
- Wan, L.; Lee, C.-C.; Hsu, C.-M.; Hwu, W.-L.; Yang, C.-C.; Tsai, C.-H.; Tsai, F.-J. Identification of Eight Novel Mutations of the Acid α-Glucosidase Gene Causing the Infantile or Juvenile Form of Glycogen Storage Disease Type II. J. Neurol. 2008, 255, 831–838. [Google Scholar] [CrossRef]
- Bellomo, F.; De Leo, E.; Taranta, A.; Giaquinto, L.; Di Giovamberardino, G.; Montefusco, S.; Rega, L.R.; Pastore, A.; Medina, D.L.; Di Bernardo, D.; et al. Drug Repurposing in Rare Diseases: An Integrative Study of Drug Screening and Transcriptomic Analysis in Nephropathic Cystinosis. Int. J. Mol. Sci. 2021, 22, 12829. [Google Scholar] [CrossRef] [PubMed]
- Mamouei, Z.; Alqarihi, A.; Singh, S.; Xu, S.; Mansour, M.K.; Ibrahim, A.S.; Uppuluri, P. Alexidine Dihydrochloride Has Broad-Spectrum Activities against Diverse Fungal Pathogens. mSphere 2018, 3, e00539-18. [Google Scholar] [CrossRef]
- Doughty-Shenton, D.; Joseph, J.D.; Zhang, J.; Pagliarini, D.J.; Kim, Y.; Lu, D.; Dixon, J.E.; Casey, P.J. Pharmacological Targeting of the Mitochondrial Phosphatase PTPMT1. J. Pharmacol. Exp. Ther. 2010, 333, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Koziak, K.; Kowalewska, M.; Zegrocka-Stendel, O.; Dutkiewicz, M.; Maciejko, D.; Domanski, D.; Perzanowska, A.; Fogtman, A.; Grabowska, I. The Cellular and Molecular Basis for Therapeutic Effectiveness of β-Escin. Atherosclerosis 2014, 235, e267. [Google Scholar] [CrossRef]
- Watanabe, Y.; Okumura, K.; Hashimoto, H.; Ito, T.; Ogawa, K.; Satake, T. Effects of Digoxin on Acetylcholine and Norepinephrine Concentrations in Rat Myocardium. J. Cardiovasc. Pharmacol. 1989, 13, 702–708. [Google Scholar] [CrossRef]
- Chick, J.; Gough, K.; Falkowski, W.; Kershaw, P.; Hore, B.; Mehta, B.; Ritson, B.; Ropner, R.; Torley, D. Disulfiram Treatment of Alcoholism. Br. J. Psychiatry 1992, 161, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.A.; Niemegeers, C.J.; Schellekens, K.H.; Lenaerts, F.M.; Verbruggen, F.J.; van Nueten, J.M.; Marsboom, R.H.; Hérin, V.V.; Schaper, W.K. The Pharmacology of Fluspirilene (R 6218), a Potent, Long-Acting and Injectable Neuroleptic Drug. Arzneimittelforschung 1970, 20, 1689–1698. [Google Scholar] [PubMed]
- De Leo, E.; Elmonem, M.A.; Berlingerio, S.P.; Berquez, M.; Festa, B.P.; Raso, R.; Bellomo, F.; Starborg, T.; Janssen, M.J.; Abbaszadeh, Z.; et al. Cell-Based Phenotypic Drug Screening Identifies Luteolin as Candidate Therapeutic for Nephropathic Cystinosis. J. Am. Soc. Nephrol. JASN 2020, 31, 1522–1537. [Google Scholar] [CrossRef] [PubMed]
- Harborne, J.B.; Williams, C.A. Advances in Flavonoid Research since 1992. Phytochemistry 2000, 55, 481–504. [Google Scholar] [CrossRef]
- Lin, Y.; Shi, R.; Wang, X.; Shen, H.-M. Luteolin, a Flavonoid with Potential for Cancer Prevention and Therapy. Curr. Cancer Drug Targets 2008, 8, 634–646. [Google Scholar] [CrossRef]
- Masten, M.C.; Mink, J.W.; Augustine, E.F. Batten Disease: An Expert Update on Agents in Preclinical and Clinical Trials. Expert Opin. Investig. Drugs 2020, 29, 1317–1322. [Google Scholar] [CrossRef]
- Soldati, C.; Lopez-Fabuel, I.; Wanderlingh, L.G.; Garcia-Macia, M.; Monfregola, J.; Esposito, A.; Napolitano, G.; Guevara-Ferrer, M.; Scotto Rosato, A.; Krogsaeter, E.K.; et al. Repurposing of Tamoxifen Ameliorates CLN3 and CLN7 Disease Phenotype. EMBO Mol. Med. 2021, 13, e13742. [Google Scholar] [CrossRef]
- Centa, J.L.; Stratton, M.P.; Pratt, M.A.; Osterlund Oltmanns, J.R.; Wallace, D.G.; Miller, S.A.; Weimer, J.M.; Hastings, M.L. Protracted CLN3 Batten Disease in Mice That Genetically Model an Exon-Skipping Therapeutic Approach. Mol. Ther. Nucleic Acids 2023, 33, 15–27. [Google Scholar] [CrossRef]
- Heins-Marroquin, U.; Singh, R.R.; Perathoner, S.; Gavotto, F.; Ruiz, C.M.; Patraskaki, M.; Gomez-Giro, G.; Borgmann, F.K.; Meyer, M.; Carpentier, A.; et al. CLN3 Deficiency Leads to Neurological and Metabolic Perturbations during Early Development. Life Sci. Alliance 2024, 7, e202302057. [Google Scholar] [CrossRef]
- Gayi, E.; Neff, L.A.; Massana Muñoz, X.; Ismail, H.M.; Sierra, M.; Mercier, T.; Décosterd, L.A.; Laporte, J.; Cowling, B.S.; Dorchies, O.M.; et al. Tamoxifen Prolongs Survival and Alleviates Symptoms in Mice with Fatal X-Linked Myotubular Myopathy. Nat. Commun. 2018, 9, 4848. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Spampanato, C.; Feeney, E.; Li, L.; Cardone, M.; Lim, J.; Annunziata, F.; Zare, H.; Polishchuk, R.; Puertollano, R.; Parenti, G.; et al. Transcription Factor EB (TFEB) Is a New Therapeutic Target for Pompe Disease. EMBO Mol. Med. 2013, 5, 691–706. [Google Scholar] [CrossRef]
- Palmer, D.N. The Relevance of the Storage of Subunit c of ATP Synthase in Different Forms and Models of Batten Disease (NCLs). Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2287–2291. [Google Scholar] [CrossRef]
- Kauss, V.; Dambrova, M.; Medina, D.L. Pharmacological Approaches to Tackle NCLs. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165553. [Google Scholar] [CrossRef] [PubMed]
- Capuozzo, A.; Montefusco, S.; Cacace, V.; Sofia, M.; Esposito, A.; Napolitano, G.; Nusco, E.; Polishchuk, E.; Pizzo, M.T.; De Risi, M.; et al. Fluoxetine Ameliorates Mucopolysaccharidosis Type IIIA. Mol. Ther. 2022, 30, 1432–1450. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, L.; Torres-Sanchez, S.; Bravo, L.; Mico, J.A.; Berrocoso, E. Fluoxetine: A Case History of Its Discovery and Preclinical Development. Expert Opin. Drug Discov. 2014, 9, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Schlotawa, L.; Tyka, K.; Kettwig, M.; Ahrens-Nicklas, R.C.; Baud, M.; Berulava, T.; Brunetti-Pierri, N.; Gagne, A.; Herbst, Z.M.; Maguire, J.A.; et al. Drug Screening Identifies Tazarotene and Bexarotene as Therapeutic Agents in Multiple Sulfatase Deficiency. EMBO Mol. Med. 2023, 15, e14837. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Hay Mele, B.; Cozzolino, F.; Monaco, V.; Cimmaruta, C.; Monti, M.; Andreotti, G.; Monticelli, M. Enzyme Replacement Therapy for FABRY Disease: Possible Strategies to Improve Its Efficacy. Int. J. Mol. Sci. 2023, 24, 4548. [Google Scholar] [CrossRef]
- Bragato, C.; Blasevich, F.; Ingenito, G.; Mantegazza, R.; Maggi, L. Therapeutic Efficacy of 3,4-Diaminopyridine Phosphate on Neuromuscular Junction in Pompe Disease. Biomed. Pharmacother. 2021, 137, 111357. [Google Scholar] [CrossRef]
- Bonanno, S.; Pasanisi, M.B.; Frangiamore, R.; Maggi, L.; Antozzi, C.; Andreetta, F.; Campanella, A.; Brenna, G.; Cottini, L.; Mantegazza, R. Amifampridine Phosphate in the Treatment of Muscle-Specific Kinase Myasthenia Gravis: A Phase IIb, Randomized, Double-Blind, Placebo-Controlled, Double Crossover Study. SAGE Open Med. 2018, 6, 205031211881901. [Google Scholar] [CrossRef] [PubMed]
- LeVine, S.M.; Tsau, S. Substrate Reduction Therapy for Krabbe Disease: Exploring the Repurposing of the Antibiotic D-Cycloserine. Front. Pediatr. 2022, 9, 807973. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Freedman, J.E. Dipyridamole, Cerebrovascular Disease, and the Vasculature. Vasc. Pharmacol. 2008, 48, 143–149. [Google Scholar] [CrossRef]
- Schade, S.; Paulus, W. D-Cycloserine in Neuropsychiatric Diseases: A Systematic Review. Int. J. Neuropsychopharmacol. 2016, 19, pyv102. [Google Scholar] [CrossRef] [PubMed]
- Malerba, M.; Ragnoli, B. Ambroxol in the 21st Century: Pharmacological and Clinical Update. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1119–1129. [Google Scholar] [CrossRef]
- Zimran, A.; Altarescu, G.; Elstein, D. Pilot Study Using Ambroxol as a Pharmacological Chaperone in Type 1 Gaucher Disease. Blood Cells Mol. Dis. 2013, 50, 134–137. [Google Scholar] [CrossRef]
- Pantoom, S.; Hules, L.; Schöll, C.; Petrosyan, A.; Monticelli, M.; Pospech, J.; Cubellis, M.V.; Hermann, A.; Lukas, J. Mechanistic Insight into the Mode of Action of Acid β-Glucosidase Enhancer Ambroxol. Int. J. Mol. Sci. 2022, 23, 3536. [Google Scholar] [CrossRef]
- Pavlova, E.V.; Lev, D.; Michelson, M.; Yosovich, K.; Michaeli, H.G.; Bright, N.A.; Manna, P.T.; Dickson, V.K.; Tylee, K.L.; Church, H.J.; et al. Juvenile Mucopolysaccharidosis plus Disease Caused by a Missense Mutation in VPS33A. Hum. Mutat. 2022, 43, 2265–2278. [Google Scholar] [CrossRef]
- Shayman, J.A. Eliglustat Tartrate. Drugs Future 2010, 35, 613. [Google Scholar] [CrossRef]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Zhou, S.; Xu, S.; Yan, Y.; Yu, H.; Ling, S.; Luo, J. Decreased Purinergic Inhibition of Synaptic Activity in a Mouse Model of Niemann-Pick Disease Type C. Hippocampus 2011, 21, 212–219. [Google Scholar] [CrossRef]
- Pepponi, R.; De Simone, R.; De Nuccio, C.; Visentin, S.; Matteucci, A.; Bernardo, A.; Popoli, P.; Ferrante, A. Repurposing Dipyridamole in Niemann Pick Type C Disease: A Proof of Concept Study. Int. J. Mol. Sci. 2022, 23, 3456. [Google Scholar] [CrossRef]
- Braun, F.; Abed, A.; Sellung, D.; Rogg, M.; Woidy, M.; Eikrem, O.; Wanner, N.; Gambardella, J.; Laufer, S.D.; Haas, F.; et al. Accumulation of α-Synuclein Mediates Podocyte Injury in Fabry Nephropathy. J. Clin. Investig. 2023, 133, e157782. [Google Scholar] [CrossRef] [PubMed]
- Morales, C.; Fernandez, M.; Ferrer, R.; Raimunda, D.; Carrer, D.C.; Bollo, M. Ursodeoxycholic Acid Binds PERK and Ameliorates Neurite Atrophy in a Cellular Model of GM2 Gangliosidosis. Int. J. Mol. Sci. 2023, 24, 7209. [Google Scholar] [CrossRef] [PubMed]
- Cougnoux, A.; Yerger, J.C.; Fellmeth, M.; Serra-Vinardell, J.; Navid, F.; Wassif, C.A.; Cawley, N.X.; Porter, F.D. Reduction of Glutamate Neurotoxicity: A Novel Therapeutic Approach for Niemann-Pick Disease, Type C1. Mol. Genet. Metab. 2021, 134, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Doble, A. The Pharmacology and Mechanism of Action of Riluzole. Neurology 1996, 47, 233S–241S. [Google Scholar] [CrossRef]
- Delaleu, N.; Marti, H.-P.; Strauss, P.; Sekulic, M.; Osman, T.; Tøndel, C.; Skrunes, R.; Leh, S.; Svarstad, E.; Nowak, A.; et al. Systems Analyses of the Fabry Kidney Transcriptome and Its Response to Enzyme Replacement Therapy Identified and Cross-Validated Enzyme Replacement Therapy-Resistant Targets Amenable to Drug Repurposing. Kidney Int. 2023, 104, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Björkhem, I.; Diczfalusy, U. Oxysterols: Friends, Foes, or Just Fellow Passengers? Arterioscler. Thromb. Vasc. Biol. 2002, 22, 734–742. [Google Scholar] [CrossRef]
- Ohgane, K.; Karaki, F.; Noguchi-Yachide, T.; Dodo, K.; Hashimoto, Y. Structure–Activity Relationships of Oxysterol-Derived Pharmacological Chaperones for Niemann–Pick Type C1 Protein. Bioorganic Med. Chem. Lett. 2014, 24, 3480–3485. [Google Scholar] [CrossRef] [PubMed]
- Ryno, L.M.; Wiseman, R.L.; Kelly, J.W. Targeting Unfolded Protein Response Signaling Pathways to Ameliorate Protein Misfolding Diseases. Curr. Opin. Chem. Biol. 2013, 17, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, M.; Liguori, L.; Allocca, M.; Bosso, A.; Andreotti, G.; Lukas, J.; Monti, M.C.; Morretta, E.; Cubellis, M.V.; Hay Mele, B. Drug Repositioning for Fabry Disease: Acetylsalicylic Acid Potentiates the Stabilization of Lysosomal α-Galactosidase by Pharmacological Chaperones. Int. J. Mol. Sci. 2022, 23, 5105. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, M.; Hay Mele, B.; Allocca, M.; Liguori, L.; Lukas, J.; Monti, M.C.; Morretta, E.; Cubellis, M.V.; Andreotti, G. Curcumin Has Beneficial Effects on Lysosomal α-Galactosidase: Potential Implications for the Cure of Fabry Disease. Int. J. Mol. Sci. 2023, 24, 1095. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Hou, L.; Gu, Y.; Lu, X.; Li, J.; Li, Q. Drug–Disease Association Prediction with Literature Based Multi-Feature Fusion. Front. Pharmacol. 2023, 14, 1205144. [Google Scholar] [CrossRef]
- Zhao, B.-W.; Hu, L.; You, Z.-H.; Wang, L.; Su, X.-R. HINGRL: Predicting Drug–Disease Associations with Graph Representation Learning on Heterogeneous Information Networks. Brief. Bioinform. 2022, 23, bbab515. [Google Scholar] [CrossRef]
- Moon, C.; Jin, C.; Dong, X.; Abrar, S.; Zheng, W.; Chirkova, R.Y.; Tropsha, A. Learning Drug-Disease-Target Embedding (DDTE) from Knowledge Graphs to Inform Drug Repurposing Hypotheses. J. Biomed. Inform. 2021, 119, 103838. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, J.; Liu, J.; Shi, D.; Li, X.; Wang, M.; Lu, T.; Jiang, B.; Xia, C.; Lin, H.; et al. Rapid Repurposing of Novel Combination Drugs for the Treatment of Heart Failure via a Computationally Guided Network Screening Approach. J. Chem. Inf. Model. 2022, 62, 5223–5232. [Google Scholar] [CrossRef]
- Yang, X.; Yang, G.; Chu, J. Self-Supervised Learning for Label Sparsity in Computational Drug Repositioning. IEEE/ACM Trans. Comput. Biol. Bioinf. 2023, 20, 3245–3256. [Google Scholar] [CrossRef]
- Wu, X.; Yang, L.; Gong, J.; Zhou, C.; Lin, T.; Liu, X.; Yu, P.S. Dimension Independent Mixup for Hard Negative Sample in Collaborative Filtering. In Proceedings of the 32nd ACM International Conference on Information and Knowledge Management, Birmingham, UK, 21–25 October 2023; ACM: Birmingham, UK, 2023; pp. 2785–2794. [Google Scholar]
- Esmail, S.; Danter, W.R. Artificially Induced Pluripotent Stem Cell-Derived Whole-Brain Organoid for Modelling the Pathophysiology of Metachromatic Leukodystrophy and Drug Repurposing. Biomedicines 2021, 9, 440. [Google Scholar] [CrossRef]
- Villalba Silva, G.C.; Steindorff, T.; Silvestri Schuh, R.; Cardoso Flores, N.; Matte, U. Drug Repositioning Applied to Cardiovascular Disease in Mucopolysaccharidosis. Life 2022, 12, 2085. [Google Scholar] [CrossRef] [PubMed]
- Abdelhakim, M.; McMurray, E.; Syed, A.R.; Kafkas, S.; Kamau, A.A.; Schofield, P.N.; Hoehndorf, R. DDIEM: Drug Database for Inborn Errors of Metabolism. Orphanet J. Rare Dis. 2020, 15, 146. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Malisan, F.; Condò, I.; Testi, R. Drug Repositioning in Friedreich Ataxia. Front. Neurosci. 2022, 16, 814445. [Google Scholar] [CrossRef] [PubMed]
- Vaz, E.S.; Vassiliades, S.V.; Giarolla, J.; Polli, M.C.; Parise-Filho, R. Drug Repositioning in the COVID-19 Pandemic: Fundamentals, Synthetic Routes, and Overview of Clinical Studies. Eur. J. Clin. Pharmacol. 2023, 79, 723–751. [Google Scholar] [CrossRef]
- Hernández-Parra, H.; Cortés, H.; Avalos-Fuentes, J.A.; Del Prado-Audelo, M.; Florán, B.; Leyva-Gómez, G.; Sharifi-Rad, J.; Cho, W.C. Repositioning of Drugs for Parkinson’s Disease and Pharmaceutical Nanotechnology Tools for Their Optimization. J. Nanobiotechnol. 2022, 20, 413. [Google Scholar] [CrossRef]

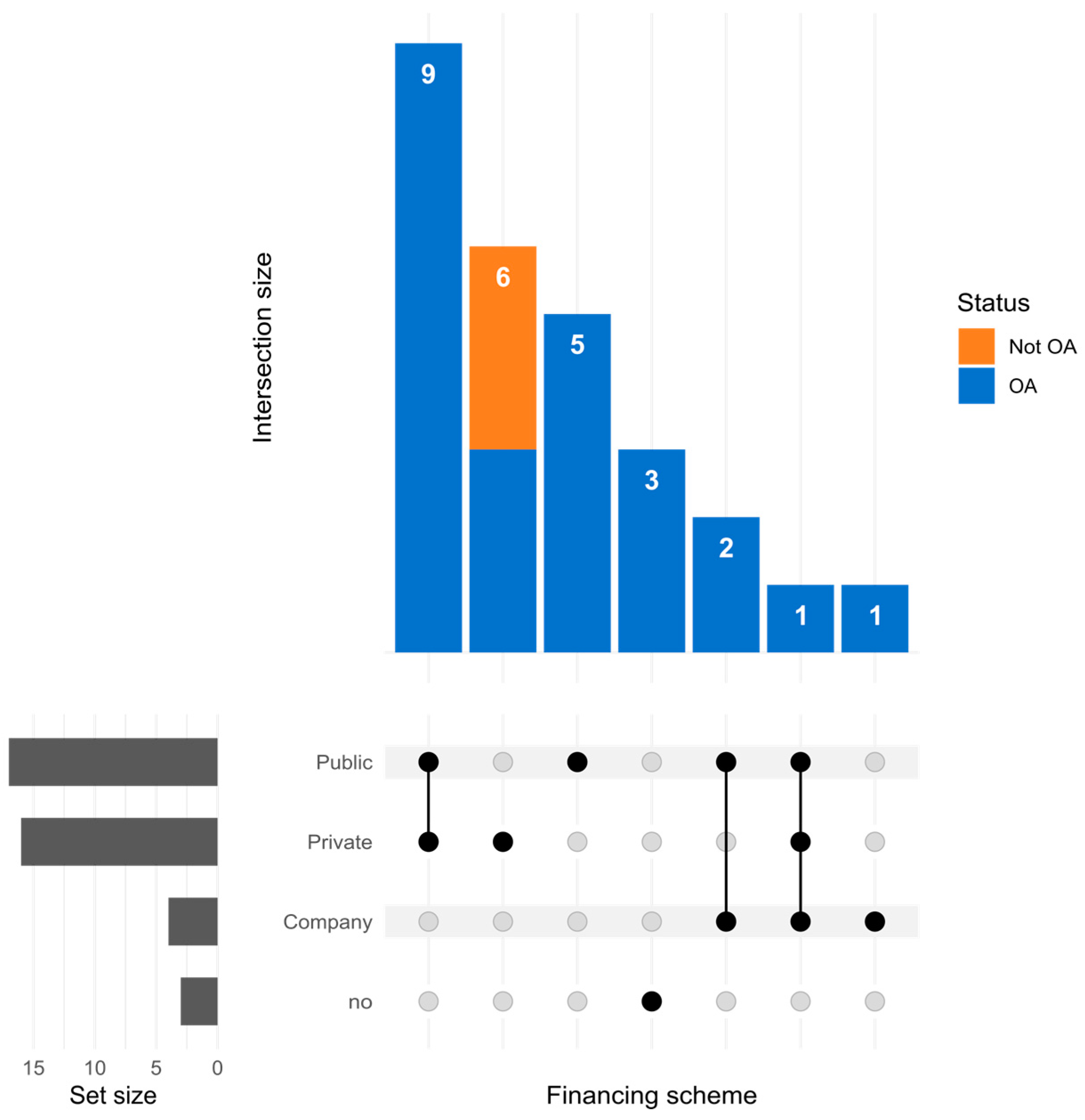
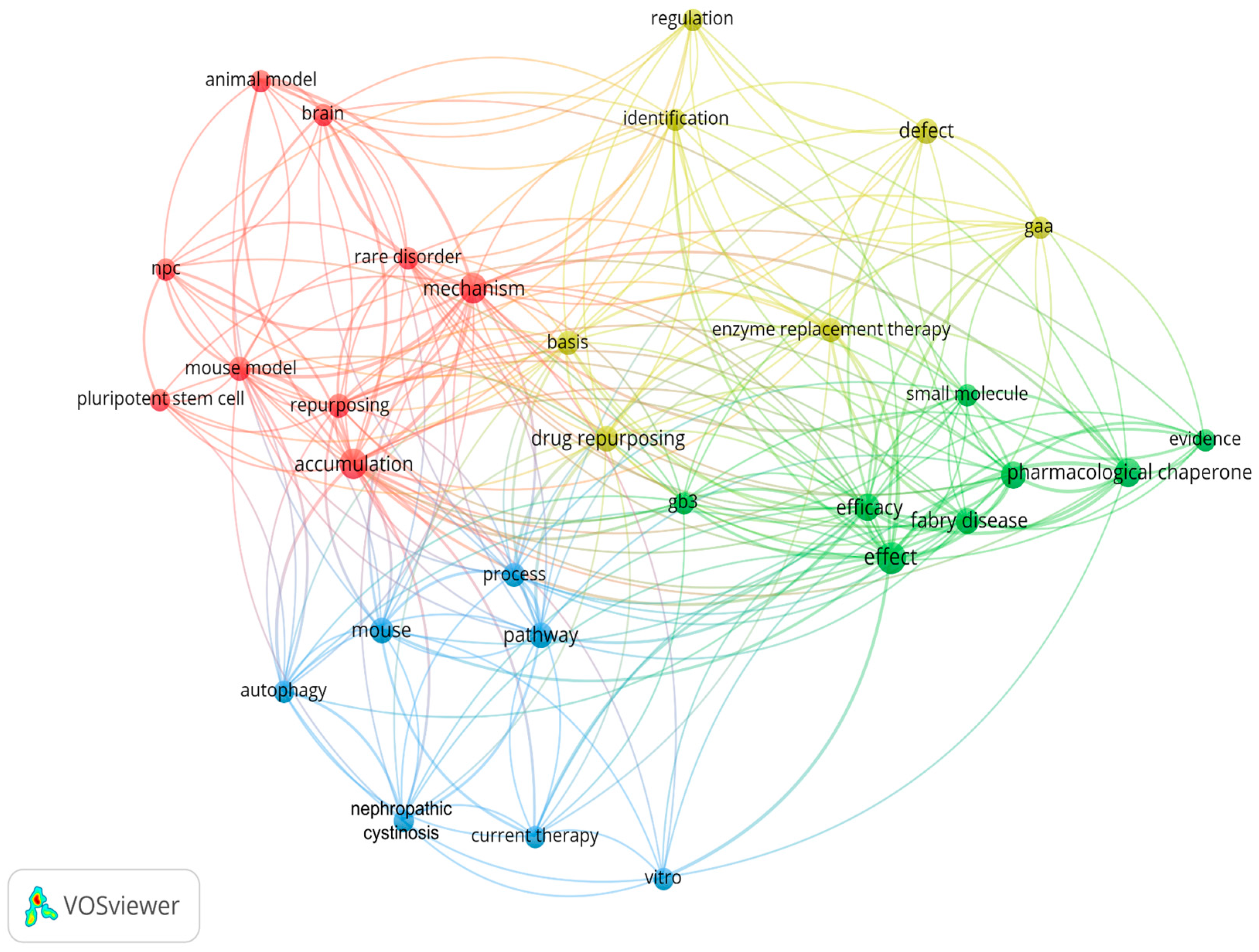

{kind=link}
{kind=link}
{kind=link}
| DOI | Synthesis |
|---|---|
| 10.3390/ijms23073536 | Ambroxol showed limited in vitro chaperone capabilities for GD, requiring further investigation into its mechanism of action. |
| 10.3390/cells10123294 | DSF demonstrated cystine-depleting and anti-apoptotic effects in nephropathic cystinosis but has dose-dependent toxicity and fails to prevent renal lesions. |
| 10.3389/fped.2021.807973 | D-cycloserine extended the lifespan of twitcher mice, suggesting potential as a therapy for KD through partial SPT inhibition and other therapeutic mechanisms. |
| 10.1016/j.biopha.2021.111357 | 3,4-DAP showed potential for treating PD-related neuromuscular transmission dysfunction, improving neuromuscular junction structure and zebrafish behavior after treatment. |
| 10.3390/ijms24087209 | UDCA reduced endoplasmic reticulum stress-induced neurite atrophy and pro-apoptotic signaling in GM2-gangliosidosis models, suggesting a therapeutic role. |
| 10.1016/j.immuni.2023.01.008 | Minocycline and etanercept protected neurons and ameliorated symptoms in a GD model by limiting microglia activation and TNF-induced neuron phagocytosis. |
| 10.3390/ijms24054548 | Combining enzyme replacement with pharmacological chaperones or targeting AGAL interactors offers the potential for optimizing FD therapy. |
| 10.1016/j.ymgme.2021.11.008 | Decreased SLC1A3 expression in NPC1 astrocytes suggests that glutamate uptake impairment contributes to disease progression; riluzole treatment improves survival. |
| 10.3390/life12122085 | Through repositioning analysis, pirfenidone and colchicine were identified as promising drugs for treating cardiovascular disease in mucopolysaccharidoses. |
| 10.3390/ijms23073456 | Increasing adenosine levels with dipyridamole reduced cholesterol accumulation and mitochondrial deficits in NPC, offering a new therapeutic approach. |
| 10.1002/humu.24479 | A study revealed a novel VPS33A mutation causing a rare mucopolysaccharidosis-plus-related variant, characterized by impaired intracellular trafficking, and suggested potential treatments using bortezomib and eliglustat to improve cellular functions. |
| 10.15252/emmm.202013742 | Research on Batten diseases identified tamoxifen as a potential treatment, showing it reduced harmful accumulation in cellular models and improved symptoms in a mouse model, highlighting the drug’s therapeutic promise. |
| 10.1016/j.kint.2023.06.029 | An analysis of Fabry nephropathy suggested that ERT can significantly reverse gene expression patterns in patients, with identified drug targets for potential treatment enhancement. |
| 10.1016/j.omtm.2020.11.011 | A screening for PD identified Defe as a drug that can restore normal gene splicing, offering a new therapeutic approach for patients with suboptimal response to current treatments. |
| 10.15252/emmm.202114837 | In MSD, tazarotene and bexarotene showed potential for correcting pathophysiology, marking a step towards developing treatments for this condition. |
| 10.1016/j.ymthe.2022.01.037 | Fluoxetine was repurposed for treating MPS IIIA, showing promise in reducing disease pathology in mouse models and suggesting a new treatment pathway through increased lysosomal exocytosis. |
| 10.3390/ijms23095105 | Research identified ASA as a potential enhancer for FD PCT, indicating a synergistic approach to treatment. |
| 10.1172/JCI157782 | A study revealed that ERT reduces globotriaosylceramide in FD but does not reverse lysosomal dysfunction, suggesting SNCA as a new therapeutic target. |
| 10.3390/ijms24021095 | Curcumin showed potential as a co-chaperone in treating FD, enhancing enzyme activity and lysosomal function in a cell model and advocating for personalized therapies. |
| 10.3390/ijms222312829 | A drug repurposing strategy for nephropathic cystinosis identified potential treatments, highlighting the approach’s effectiveness in addressing the therapeutic challenges of rare diseases. |
| 10.3390/biomedicines9040440 | The study utilized artificial brain organoids to explore MLD pathogenesis, offering new insights into potential therapeutic options through computer simulations. |
| 10.1248/CPB.C20-00194 | A novel Golgi β-galactosidase inhibitor, ARM07, showed potential in addressing LSDs and aging cell biomarkers, promising advancements in therapeutic probe development. |
| 10.1681/ASN.2019090956 | Luteolin emerged as a promising treatment for nephropathic cystinosis, enhancing autophagy-lysosome pathways and exhibiting antioxidant and anti-apoptotic properties, suggesting a potential breakthrough in renal LSD therapy. |
| 10.1016/j.bmcl.2014.05.064 | Oxysterol derivatives corrected folding defects in NPC, suggesting their utility as pharmacological chaperones for mutant NPC1 proteins, indicating a promising therapeutic strategy. |
| 10.1186/s13023-020-01428-2 | The DDIEM compiled therapeutic strategies for metabolic diseases, offering a new ontology-based knowledge base to guide treatment and management. |
| 10.1021/acsmedchemlett.0c00042 | BC was identified as a novel pharmacological chaperone for MPS IVA, showing the potential to increase enzyme activity and reduce lysosomal mass, highlighting a new therapeutic avenue. |
| 10.4155/fmc.13.123 | Chemical chaperones offer promising treatments for lysosomal storage and neurodegenerative diseases by stabilizing mutant proteins, emphasizing the potential for oral administration and blood–brain barrier penetration. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hay Mele, B.; Rossetti, F.; Cubellis, M.V.; Monticelli, M.; Andreotti, G. Drug Repurposing and Lysosomal Storage Disorders: A Trick to Treat. Genes 2024, 15, 290. https://doi.org/10.3390/genes15030290
Hay Mele B, Rossetti F, Cubellis MV, Monticelli M, Andreotti G. Drug Repurposing and Lysosomal Storage Disorders: A Trick to Treat. Genes. 2024; 15(3):290. https://doi.org/10.3390/genes15030290
Chicago/Turabian StyleHay Mele, Bruno, Federica Rossetti, Maria Vittoria Cubellis, Maria Monticelli, and Giuseppina Andreotti. 2024. "Drug Repurposing and Lysosomal Storage Disorders: A Trick to Treat" Genes 15, no. 3: 290. https://doi.org/10.3390/genes15030290