
Transcriptome Analysis of miRNA and mRNA in Porcine Skeletal Muscle following Glaesserella parasuis Challenge

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model Establishment and Tissue Sample Collection
2.2. Paraffin Section Preparation and Hematoxylin–Eosin Staining
2.3. mRNA Library Preparation and Sequencing
2.4. mRNA Raw Data Processing and Expression Analysis
2.5. miRNA Library Preparation and Sequencing
2.6. miRNA Raw Data Control and Expression Analysis
2.7. miRNA Target Gene Prediction
2.8. Functional Enrichment Analysis
2.9. Quantitative Real-Time PCR
2.10. Statistical Analyses
3. Results
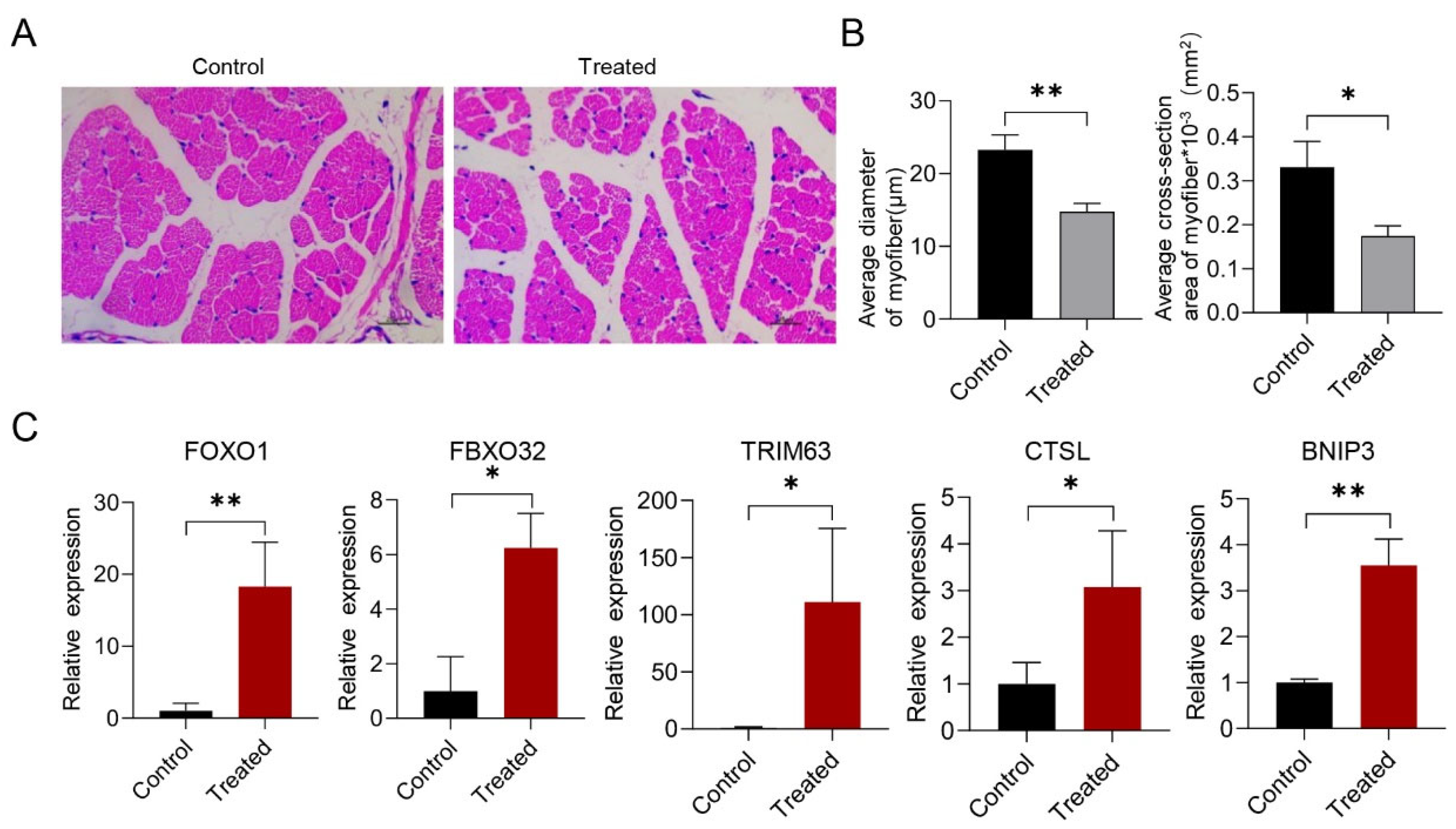
3.1. The Effect of G. parasuis Infection on Skeletal Muscle in Piglets
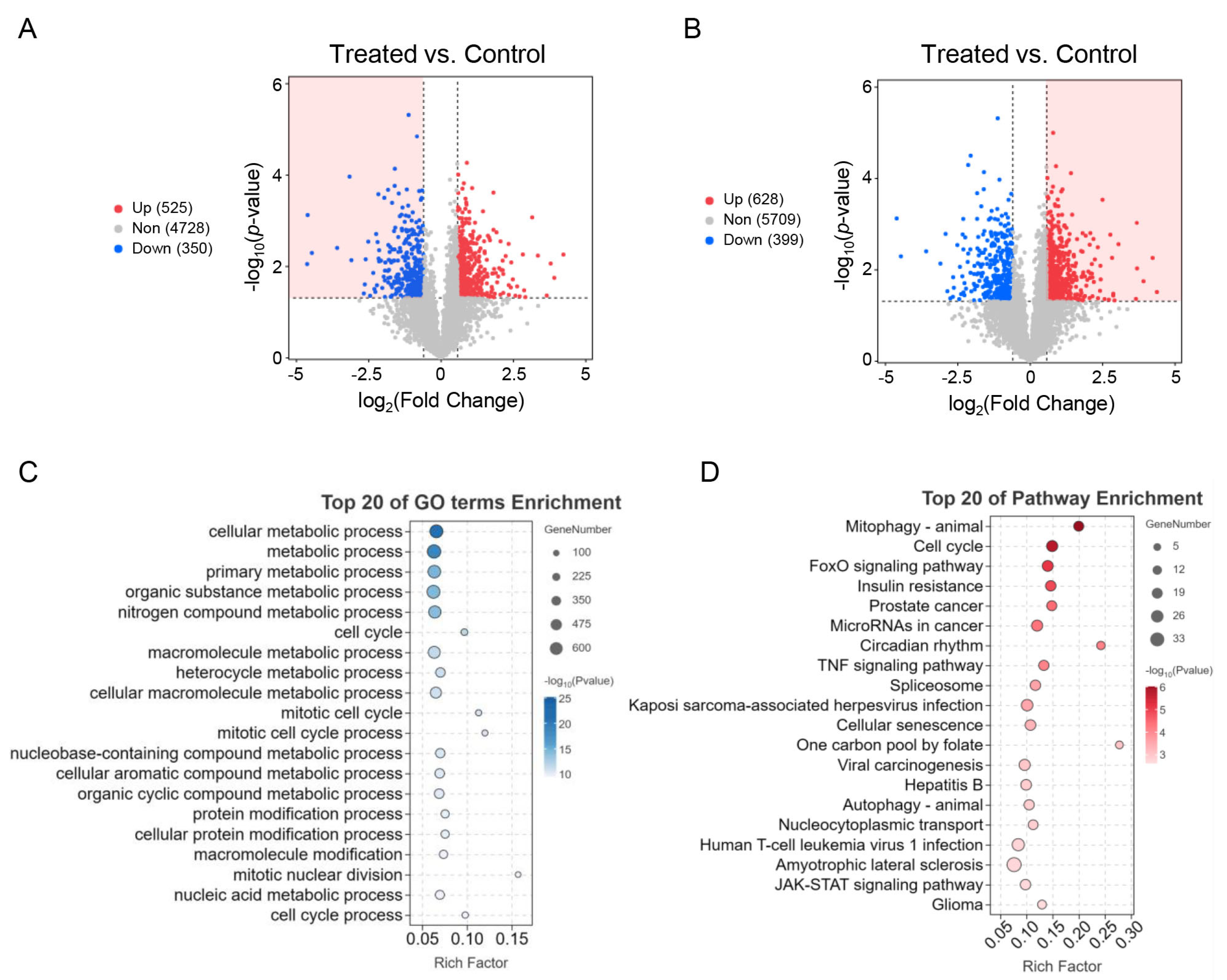
3.2. Identification of Differentially Expressed (DE) mRNAs
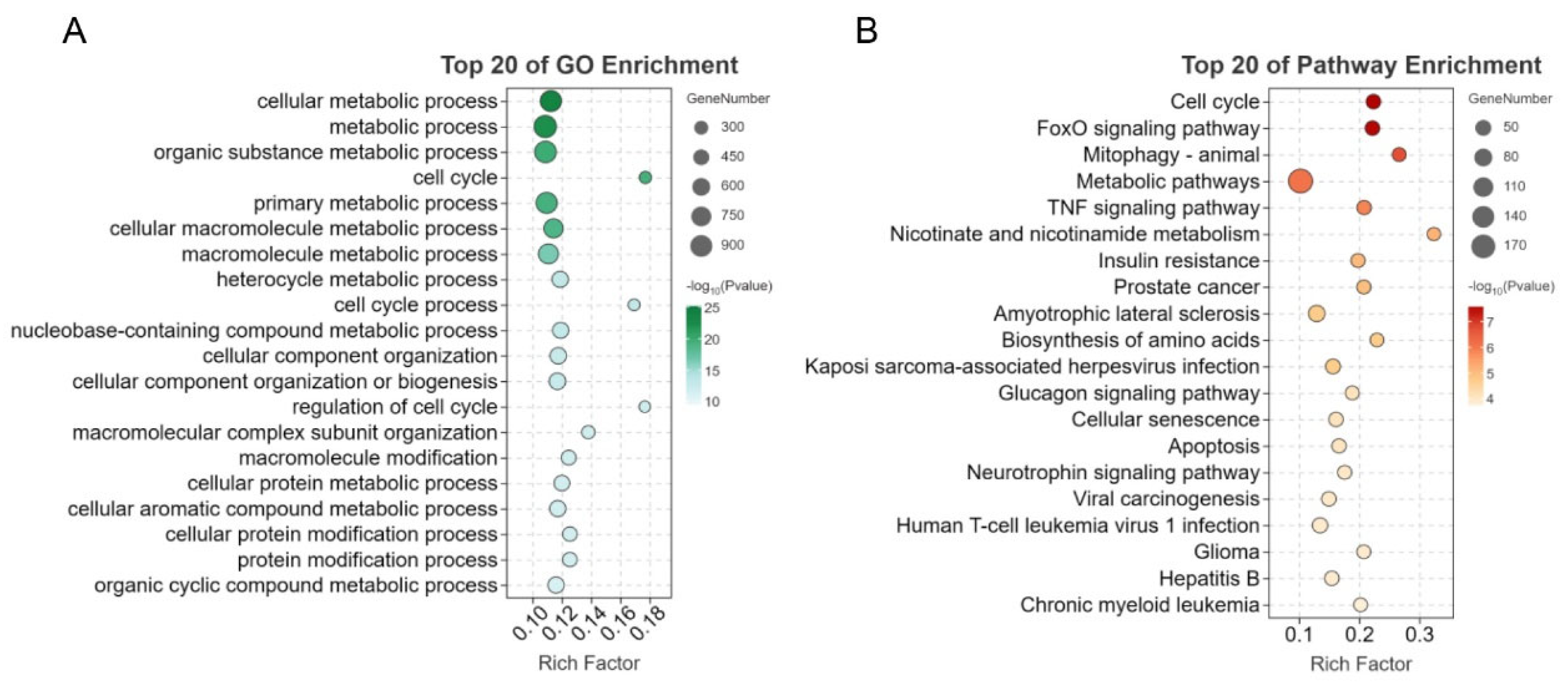
3.3. Functional Annotation of Differentially Expressed mRNAs
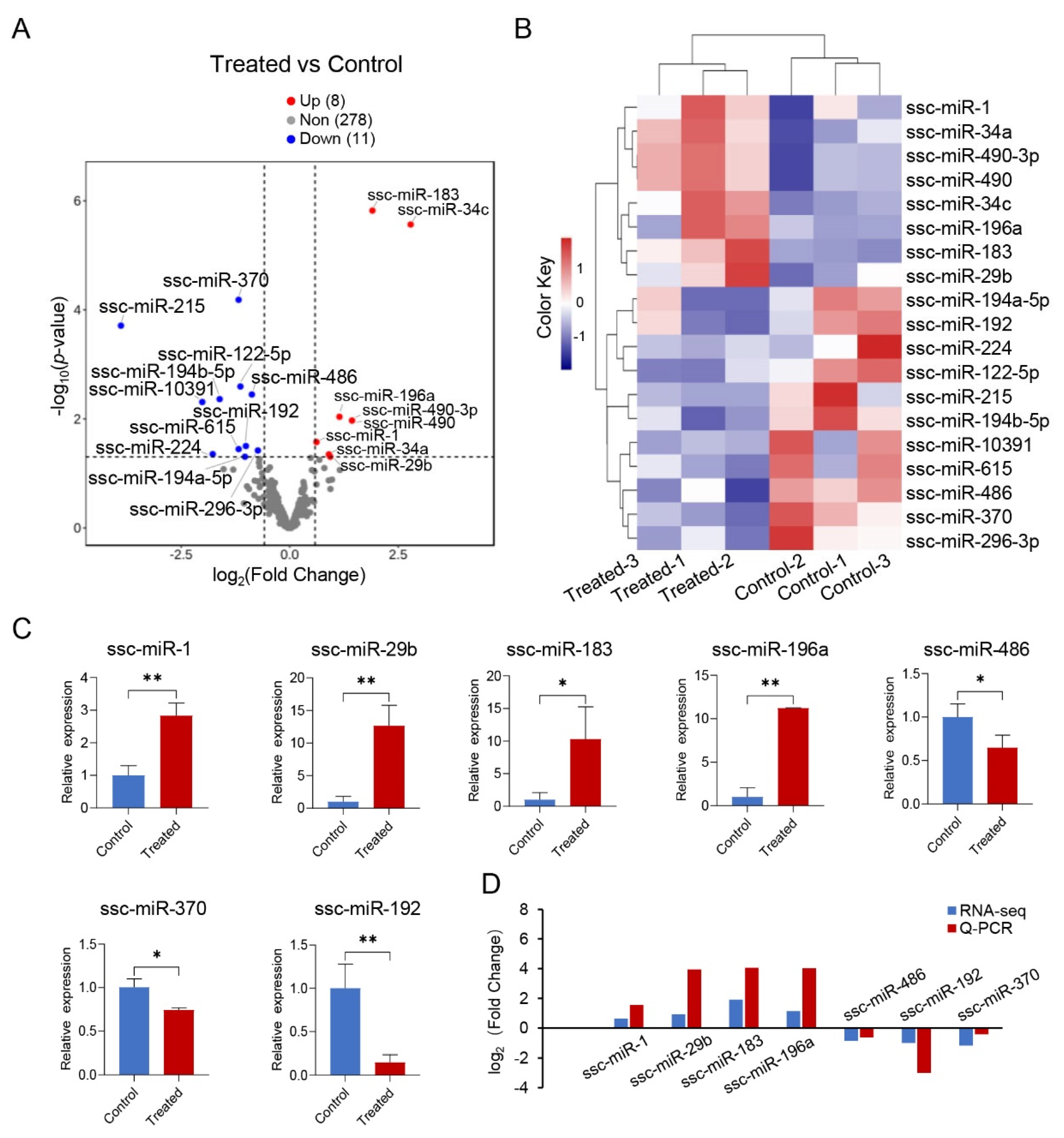
3.4. Analysis of Differentially Expressed miRNAs
3.5. Functional Enrichment Analysis of Predicted Targets of DE miRNAs
3.6. The GSEA Analysis of Expression Genes
3.7. Integrated Analysis of mRNAs and miRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dickerman, A.; Bandara, A.B.; Inzana, T.J. Phylogenomic analysis of Haemophilus parasuis and proposed reclassification to Glaesserella parasuis, gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 180–186. [Google Scholar] [CrossRef]
- Oliveira, S.; Pijoan, C. Haemophilus parasuis new trends on diagnosis, epidemiology and control. Vet. Microbiol. 2004, 99, 1–12. [Google Scholar] [CrossRef]
- Cai, X.W.; Chen, H.C.; Blackall, P.J.; Yin, Z.Y.; Wang, L.; Liu, Z.F.; Jin, M.L. Serological characterization of Haemophilus parasuis isolates from China. Vet. Microbiol. 2005, 111, 231–236. [Google Scholar] [CrossRef]
- Kielstein, P.; Rapp-Gabrielson, V.J. Designation of 15 serovars of Haemophilus parasuis on the basis of immunodiffusion using heat-stable antigen extracts. J. Clin. Microbiol. 1992, 30, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Q.; Li, J.; Lin, X.; Huang, X.; Fang, B. Epidemiology of Haemophilus parasuis isolates from pigs in China using serotyping, antimicrobial susceptibility, biofilm formation and ERIC-PCR genotyping. PeerJ 2018, 6, e5040. [Google Scholar] [CrossRef]
- Yang, X.F.; Du, Q.; Wang, X.F.; Shi, J.; Wang, T.T.; Li, P.X.; Zhong, J.H.; Tong, D.W.; Huang, Y. Porcine circovirus type 2 infection inhibits macrophage M1 polarization induced by other pathogens via viral capsid protein and host gC1qR protein. Vet. Microbiol. 2023, 285, 109871. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.X.; Pang, L.L.; Ouyang, Y.; Jiang, Y.F.; Zhang, J.J.; Qiu, Y.F.; Li, Z.J.; Li, B.B.; Liu, K.; Shao, D.H.; et al. Secondary Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus (HP-PRRSV2) Infection Augments Inflammatory Responses, Clinical Outcomes, and Pathogen Load in G.laesserella-parasuis Infected Piglets. Vet. Sci. 2023, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Mathieu-Denoncourt, A.; Letendre, C.; Auger, J.P.; Segura, M.; Aragon, V.; Lacouture, S.; Gottschalk, M. Limited Interactions between Streptococcus Suis and Haemophilus Parasuis in In Vitro Co-Infection Studies. Pathogens 2018, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, W.; Wang, Y.; Gu, C.; Liu, X.; Charreyre, C.; Fan, S.; He, Q. Coinfection with Haemophilus parasuis serovar 4 increases the virulence of porcine circovirus type 2 in piglets. Virol. J. 2017, 14, 227. [Google Scholar] [CrossRef] [PubMed]
- Palus, S.; von Haehling, S.; Springer, J. Muscle wasting: An overview of recent developments in basic research. Int. J. Cardiol. 2014, 176, 640–644. [Google Scholar] [CrossRef]
- Fang, W.Y.; Tseng, Y.T.; Lee, T.Y.; Fu, Y.C.; Chang, W.H.; Lo, W.W.; Lin, C.L.; Lo, Y.C. Triptolide prevents LPS-induced skeletal muscle atrophy via inhibiting NF-κB/TNF-α and regulating protein synthesis/degradation pathway. Br. J. Pharmacol. 2021, 178, 2998–3016. [Google Scholar] [CrossRef]
- Thomson, D.M. The Role of AMPK in the Regulation of Skeletal Muscle Size, Hypertrophy, and Regeneration. Int. J. Mol. Sci. 2018, 19, 3125. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Duquet, A.; Polesskaya, A.; Cuvellier, S.; Ait-Si-Ali, S.; Héry, P.; Pritchard, L.L.; Gerard, M.; Harel-Bellan, A. Acetylation is important for MyoD function in adult mice. EMBO Rep. 2006, 7, 1140–1146. [Google Scholar] [CrossRef]
- Rao, P.K.; Kumar, R.M.; Farkhondeh, M.; Baskerville, S.; Lodish, H.F. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc. Natl. Acad. Sci. USA 2006, 103, 8721–8726. [Google Scholar] [CrossRef]
- Liu, N.; Williams, A.H.; Kim, Y.; McAnally, J.; Bezprozvannaya, S.; Sutherland, L.B.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. USA 2007, 104, 20844–20849. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, K.K.; Buvoli, M.; Pugach, E.K.; Polmear, M.M.; Leinwand, L.A. miR-1/206 downregulates splicing factor Srsf9 to promote C2C12 differentiation. Skelet. Muscle 2019, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Liu, N.; van Rooij, E.; Olson, E.N. MicroRNA control of muscle development and disease. Curr. Opin. Cell Biol. 2009, 21, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; He, M.; Yang, L.; Zhou, R.; Yan, Y.Q.; Liang, Y.; Teng, C.B. MiR-17 and miR-19 cooperatively promote skeletal muscle cell differentiation. Cell Mol. Life Sci. 2019, 76, 5041–5054. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Q.; Wang, B.; Wu, W.; Wei, J.; Li, P.; Huang, R. miR-22 regulates C2C12 myoblast proliferation and differentiation by targeting TGFBR1. Eur. J. Cell Biol. 2018, 97, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Hu, Z.; Klein, J.D.; Zhang, L.; Fang, F.; Mitch, W.E. Decreased miR-29 suppresses myogenesis in CKD. J. Am. Soc. Nephrol. 2011, 22, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.; Mastroyiannopoulos, N.P.; Uney, J.B.; Phylactou, L.A. miR-186 inhibits muscle cell differentiation through myogenin regulation. J. Biol. Chem. 2014, 289, 3923–3935. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Du, J.; Shen, L.; Tan, Z.; Jiang, D.; Jiang, A.; Li, Q.; Tang, G.; Jiang, Y.; Wang, J.; et al. MiR-204-5p regulates C2C12 myoblast differentiation by targeting MEF2C and ERRγ. Biomed. Pharmacother. 2018, 101, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wang, J.; Sun, Y.; Zhao, T.; Luo, X.; Lu, J.; Hou, W.; Yu, X.; Xue, L.; Yan, Y.; et al. MiR-222-3p suppresses C2C12 myoblast proliferation and differentiation via the inhibition of IRS-1/PI3K/Akt pathway. J. Cell Biochem. 2023, 124, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Dhuppar, S.; Murugaiyan, G. miRNA effects on gut homeostasis: Therapeutic implications for inflammatory bowel disease. Trends Immunol. 2022, 43, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, L.; Wang, X.; Xia, L.; Zhang, Y. Advances in the role and mechanism of miRNA in inflammatory pain. Biomed. Pharmacother. 2023, 161, 114463. [Google Scholar] [CrossRef]
- Essandoh, K.; Li, Y.; Huo, J.; Fan, G.C. MiRNA-Mediated Macrophage Polarization and its Potential Role in the Regulation of Inflammatory Response. Shock 2016, 46, 122–131. [Google Scholar] [CrossRef]
- Luo, Z.; Qi, B.; Sun, Y.; Chen, Y.; Lin, J.; Qin, H.; Wang, N.; Shi, R.; Shang, X.; Chen, S.; et al. Engineering Bioactive M2 Macrophage-Polarized, Anti-inflammatory, miRNA-Based Liposomes for Functional Muscle Repair: From Exosomal Mechanisms to Biomaterials. Small 2022, 18, e2201957. [Google Scholar] [CrossRef]
- Eisenberg, I.; Eran, A.; Nishino, I.; Moggio, M.; Lamperti, C.; Amato, A.A.; Lidov, H.G.; Kang, P.B.; North, K.N.; Mitrani-Rosenbaum, S.; et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 17016–17021. [Google Scholar] [CrossRef]
- Vignier, N.; Amor, F.; Fogel, P.; Duvallet, A.; Poupiot, J.; Charrier, S.; Arock, M.; Montus, M.; Nelson, I.; Richard, I.; et al. Distinctive serum miRNA profile in mouse models of striated muscular pathologies. PLoS ONE 2013, 8, e55281. [Google Scholar] [CrossRef]
- Muñoz-Braceras, S.; Pinal-Fernandez, I.; Casal-Dominguez, M.; Pak, K.; Milisenda, J.C.; Lu, S.; Gadina, M.; Naz, F.; Gutierrez-Cruz, G.; Dell’Orso, S.; et al. Identification of Unique microRNA Profiles in Different Types of Idiopathic Inflammatory Myopathy. Cells 2023, 12, 2198. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, G.; Wan, G.; Li, M.; Chang, B.; Yi, X. Idiopathic inflammatory myopathy and non-coding RNA. Front. Immunol. 2023, 14, 1227945. [Google Scholar] [CrossRef]
- Hu, J.; Kong, M.; Ye, Y.; Hong, S.; Cheng, L.; Jiang, L. Serum miR-206 and other muscle-specific microRNAs as non-invasive biomarkers for Duchenne muscular dystrophy. J. Neurochem. 2014, 129, 877–883. [Google Scholar] [CrossRef]
- Zhang, A.; Li, M.; Wang, B.; Klein, J.D.; Price, S.R.; Wang, X.H. miRNA-23a/27a attenuates muscle atrophy and renal fibrosis through muscle-kidney crosstalk. J. Cachexia Sarcopenia Muscle 2018, 9, 755–770. [Google Scholar] [CrossRef]
- McCormack, N.M.; Calabrese, K.A.; Sun, C.M.; Tully, C.B.; Heier, C.R.; Fiorillo, A.A. Deletion of miR-146a enhances therapeutic protein restoration in model of dystrophin exon skipping. bioRxiv 2023. [Google Scholar] [CrossRef]
- Li, J.; Chan, M.C.; Yu, Y.; Bei, Y.; Chen, P.; Zhou, Q.; Cheng, L.; Chen, L.; Ziegler, O.; Rowe, G.C.; et al. miR-29b contributes to multiple types of muscle atrophy. Nat. Commun. 2017, 8, 15201. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.F.; Zhu, Y.; Zhang, S.; Liu, Q.; Zhang, J.; Zhou, H.H.; Zhou, A.; Shing, L.Y.; Chen, H.B. The model of Glaesserella parasuis infection in fasting colostrum CD piglets andconstruction of infection-related phenotype group. Chin. J. Anim. Sci. 2022, 58, 125–135. [Google Scholar] [CrossRef]
- Chen, H.B.; Cheng, L.; Xiang, M.; Zhao, S.H.; Hou, Y.Q.; Li, W.M. Establishment of Experimental Model for Colostrum-deprived Piglets Infected by Hamophilus Parasuis. Acta Ecol. Anim. Domastici 2015, 36, 53–62. [Google Scholar]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Riley, M.G.; Russell, E.G.; Callinan, R.B. Haemophilus parasuis infection in swine. J. Am. Vet. Med. Assoc. 1977, 171, 649–651. [Google Scholar]
- Palzer, A.; Austin-Busse, R.L.; Ladinig, A.; Balka, G.; Zoels, S.; Ritzmann, M. Histopathologic lesions in conventional pigs experimentally infected with Haemophilus parasuis serovar 5. Tierarztl. Prax. Ausg. G. Grosstiere Nutztiere 2015, 43, 91–96. [Google Scholar] [CrossRef]
- Guo, L.; Cheng, H.; Fu, S.; Liu, J.; Zhang, Y.; Qiu, Y.; Chen, H. Methylome and Transcriptome-Based Integration Analysis Identified Molecular Signatures Associated With Meningitis Induced by Glaesserella parasuis. Front. Immunol. 2022, 13, 840399. [Google Scholar] [CrossRef] [PubMed]
- Vahle, J.L.; Haynes, J.S.; Andrews, J.J. Experimental reproduction of Haemophilus parasuis infection in swine: Clinical, bacteriological, and morphologic findings. J. Vet. Diagn. Investig. 1995, 7, 476–480. [Google Scholar] [CrossRef]
- Hoefling, D.C. Acute myositis associated with Hemophilus parasuis in primary SPF sows. J. Vet. Diagn. Investig. 1991, 3, 354–355. [Google Scholar] [CrossRef]
- Zhang, B.; He, Y.; Xu, C.; Xu, L.; Feng, S.; Liao, M.; Ren, T. Cytolethal distending toxin (CDT) of the Haemophilus parasuis SC096 strain contributes to serum resistance and adherence to and invasion of PK-15 and PUVEC cells. Vet. Microbiol. 2012, 157, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Frandoloso, R.; Pivato, M.; Martínez-Martínez, S.; Rodríguez-Ferri, E.F.; Kreutz, L.C.; Martín, C.B. Differences in Haemophilus parasuis adherence to and invasion of AOC-45 porcine aorta endothelial cells. BMC Vet. Res. 2013, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Vanier, G.; Szczotka, A.; Friedl, P.; Lacouture, S.; Jacques, M.; Gottschalk, M. Haemophilus parasuis invades porcine brain microvascular endothelial cells. Microbiology 2006, 152, 135–142. [Google Scholar] [CrossRef]
- Frandoloso, R.; Martinez-Martinez, S.; Gutierrez-Martin, C.B.; Rodriguez-Ferri, E.F. Haemophilus parasuis serovar 5 Nagasaki strain adheres and invades PK-15 cells. Vet. Microbiol. 2012, 154, 347–352. [Google Scholar] [CrossRef]
- Auger, E.; Deslandes, V.; Ramjeet, M.; Contreras, I.; Nash, J.H.; Harel, J.; Gottschalk, M.; Olivier, M.; Jacques, M. Host-pathogen interactions of Actinobacillus pleuropneumoniae with porcine lung and tracheal epithelial cells. Infect. Immun. 2009, 77, 1426–1441. [Google Scholar] [CrossRef]
- Kang, P.; Huang, X.; Wan, Z.; Liang, T.; Wang, Y.; Li, X.; Zhang, J.; Zhu, H.; Liu, Y. Kinetics of changes in gene and microRNA expression related with muscle inflammation and protein degradation following LPS-challenge in weaned piglets. Innate Immun. 2021, 27, 23–30. [Google Scholar] [CrossRef]
- Orea-Soufi, A.; Paik, J.; Bragança, J.; Donlon, T.A.; Willcox, B.J.; Link, W. FOXO transcription factors as therapeutic targets in human diseases. Trends Pharmacol. Sci. 2022, 43, 1070–1084. [Google Scholar] [CrossRef]
- Chen, K.; Gao, P.; Li, Z.; Dai, A.; Yang, M.; Chen, S.; Su, J.; Deng, Z.; Li, L. Forkhead Box O Signaling Pathway in Skeletal Muscle Atrophy. Am. J. Pathol. 2022, 192, 1648–1657. [Google Scholar] [CrossRef]
- Crossland, H.; Constantin-Teodosiu, D.; Gardiner, S.M.; Constantin, D.; Greenhaff, P.L. A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J. Physiol. 2008, 586, 5589–5600. [Google Scholar] [CrossRef]
- Reed, S.A.; Sandesara, P.B.; Senf, S.M.; Judge, A.R. Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J. 2012, 26, 987–1000. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kamei, Y.; Sugita, S.; Akaike, F.; Kanai, S.; Miura, S.; Hirata, Y.; Troen, B.R.; Kitamura, T.; Nishino, I.; et al. The cathepsin L gene is a direct target of FOXO1 in skeletal muscle. Biochem. J. 2010, 427, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Kamei, Y.; Miura, S.; Suzuki, M.; Kai, Y.; Mizukami, J.; Taniguchi, T.; Mochida, K.; Hata, T.; Matsuda, J.; Aburatani, H.; et al. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated Type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J. Biol. Chem. 2004, 279, 41114–41123. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Yang, Z.; Liu, C.W.; Wang, R.; Tien, P.; Dale, R.; Sun, L.Q. Effect of RNA oligonucleotide targeting Foxo-1 on muscle growth in normal and cancer cachexia mice. Cancer Gene Ther. 2007, 14, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Goldberg, A.L. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J. Biol. Chem. 2013, 288, 30515–30526. [Google Scholar] [CrossRef] [PubMed]
- Mandai, S.; Mori, T.; Nomura, N.; Furusho, T.; Arai, Y.; Kikuchi, H.; Sasaki, E.; Sohara, E.; Rai, T.; Uchida, S. WNK1 regulates skeletal muscle cell hypertrophy by modulating the nuclear localization and transcriptional activity of FOXO4. Sci. Rep. 2018, 8, 9101. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, R.S.; Workeneh, B.; Dong, Y.L.; Wang, X.N.; Hu, Z.Y. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 2012, 82, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Sun, H.; Zeng, W.; He, J.; Mao, X. Upregulation of MircoRNA-370 induces proliferation in human prostate cancer cells by downregulating the transcription factor FOXO1. PLoS ONE 2012, 7, e45825. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, F.; Zhao, H.; Wang, M.; Zhang, Y. Circular RNA circCHFR Facilitates the Proliferation and Migration of Vascular Smooth Muscle via miR-370/FOXO1/Cyclin D1 Pathway. Mol. Ther. Nucleic Acids 2019, 16, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, C.; Li, S.; Li, T.; Li, Y.; Wang, N.; Bao, X.; Xue, P.; Liu, S. BMSC-Derived Exosomes Inhibit Dexamethasone-Induced Muscle Atrophy via the miR-486-5p/FoxO1 Axis. Front. Endocrinol. 2021, 12, 681267. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Han, D.S.; Hsu, C.C.; Wang, J.S. Circulating MicroRNA-486 and MicroRNA-146a serve as potential biomarkers of sarcopenia in the older adults. BMC Geriatr. 2021, 21, 86. [Google Scholar] [CrossRef]
- Chang, Y.C.; Liu, H.W.; Chan, Y.C.; Hu, S.H.; Liu, M.Y.; Chang, S.J. The green tea polyphenol epigallocatechin-3-gallate attenuates age-associated muscle loss via regulation of miR-486-5p and myostatin. Arch. Biochem. Biophys. 2020, 692, 108511. [Google Scholar] [CrossRef]
- Zhang, P.; Du, J.; Guo, X.; Wu, S.; He, J.; Li, X.; Shen, L.; Chen, L.; Li, B.; Zhang, J.; et al. LncMyoD Promotes Skeletal Myogenesis and Regulates Skeletal Muscle Fiber-Type Composition by Sponging miR-370-3p. Genes 2021, 12, 589. [Google Scholar] [CrossRef]
- Siengdee, P.; Trakooljul, N.; Murani, E.; Schwerin, M.; Wimmers, K.; Ponsuksili, S. MicroRNAs Regulate Cellular ATP Levels by Targeting Mitochondrial Energy Metabolism Genes during C2C12 Myoblast Differentiation. PLoS ONE 2015, 10, e0127850. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Yang, C.Q.; Chen, Q.; Liu, J.; Zhang, G.; Dong, C.; Liu, X.L.; Farooq, H.M.U.; Zhao, S.Q.; Luo, L.H.; et al. miR-194-Loaded Gelatin Nanospheres Target MEF2C to Suppress Muscle Atrophy in a Mechanical Unloading Model. Mol. Pharm. 2021, 18, 2959–2973. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zheng, C.; Guo, Q.; Yin, Y.; Duan, Y.; Li, F. LPS-related muscle loss is associated with the alteration of Bacteroidetes abundance, systemic inflammation, and mitochondrial morphology in a weaned piglet model. Sci. China Life Sci. 2024. [Google Scholar] [CrossRef]
- Duan, Y.; Zheng, C.; Zhong, Y.; Song, B.; Yan, Z.; Kong, X.; Deng, J.; Li, F.; Yin, Y. Beta-hydroxy beta-methyl butyrate decreases muscle protein degradation via increased Akt/FoxO3a signaling and mitochondrial biogenesis in weanling piglets after lipopolysaccharide challenge. Food Funct. 2019, 10, 5152–5165. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Reads | Q30 (%) | Clean Reads | Clean Reads Ratio (%) | Mapped Reads Ratio (%) |
|---|---|---|---|---|---|
| Treated-1 | 35,530,548 | 91.75 | 34,966,632 | 98.41 | 92.42 |
| Treated-2 | 39,459,754 | 91.13 | 38,664,210 | 97.9 | 92.81 |
| Treated-3 | 33,483,658 | 90.44 | 32,747,122 | 97.80 | 91.50 |
| Control-1 | 31,488,902 | 91.33 | 31,085,022 | 98.72 | 93.97 |
| Control-2 | 40,461,752 | 90.23 | 38,798,728 | 95.89 | 90.06 |
| Control-3 | 32,952,226 | 90.66 | 32,518,392 | 98.68 | 92.97 |
| Samples | Raw Reads | Q30 (%) | Clean Reads | Clean Reads Ratio (%) | Mapped Reads Ratio (%) |
|---|---|---|---|---|---|
| Treated-1 | 9,445,249 | 92.60 | 9,332,514 | 98.81 | 84.05 |
| Treated-2 | 8,657,021 | 92.05 | 8,285,011 | 95.70 | 80.55 |
| Treated-3 | 10,129,573 | 92.40 | 9,927,882 | 98.01 | 82.03 |
| Control-1 | 9,506,066 | 92.31 | 9,343,438 | 98.29 | 82.98 |
| Control-2 | 8,975,127 | 91.70 | 8,764,166 | 97.65 | 78.74 |
| Control-3 | 8,501,670 | 92.40 | 8,153,429 | 95.90 | 81.95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Chen, X.; Deng, X.; Zhang, X.; Zeng, X.; Xu, K.; Chen, H. Transcriptome Analysis of miRNA and mRNA in Porcine Skeletal Muscle following Glaesserella parasuis Challenge. Genes 2024, 15, 359. https://doi.org/10.3390/genes15030359
Zhou H, Chen X, Deng X, Zhang X, Zeng X, Xu K, Chen H. Transcriptome Analysis of miRNA and mRNA in Porcine Skeletal Muscle following Glaesserella parasuis Challenge. Genes. 2024; 15(3):359. https://doi.org/10.3390/genes15030359
Chicago/Turabian StyleZhou, Huanhuan, Xuexue Chen, Xiangwei Deng, Xiaoyu Zhang, Xinqi Zeng, Ke Xu, and Hongbo Chen. 2024. "Transcriptome Analysis of miRNA and mRNA in Porcine Skeletal Muscle following Glaesserella parasuis Challenge" Genes 15, no. 3: 359. https://doi.org/10.3390/genes15030359