
Genome-Wide Association Study Uncovers Genomic Regions Associated with Coleoptile Length in a Worldwide Collection of Oat
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Phenotype Evaluation
2.3. Statistical Analysis
2.4. Genomic Data Analysis
2.5. Association Analysis
2.6. Comparative Mapping
2.7. Effects of Favorable Alleles
2.8. Putative Candidate Gene Analysis
3. Results
3.1. Coleoptile Length in Oat Accession
3.2. Genome-Wide Association Analysis of Coleoptile Length
3.3. Comparison to Previous Studies
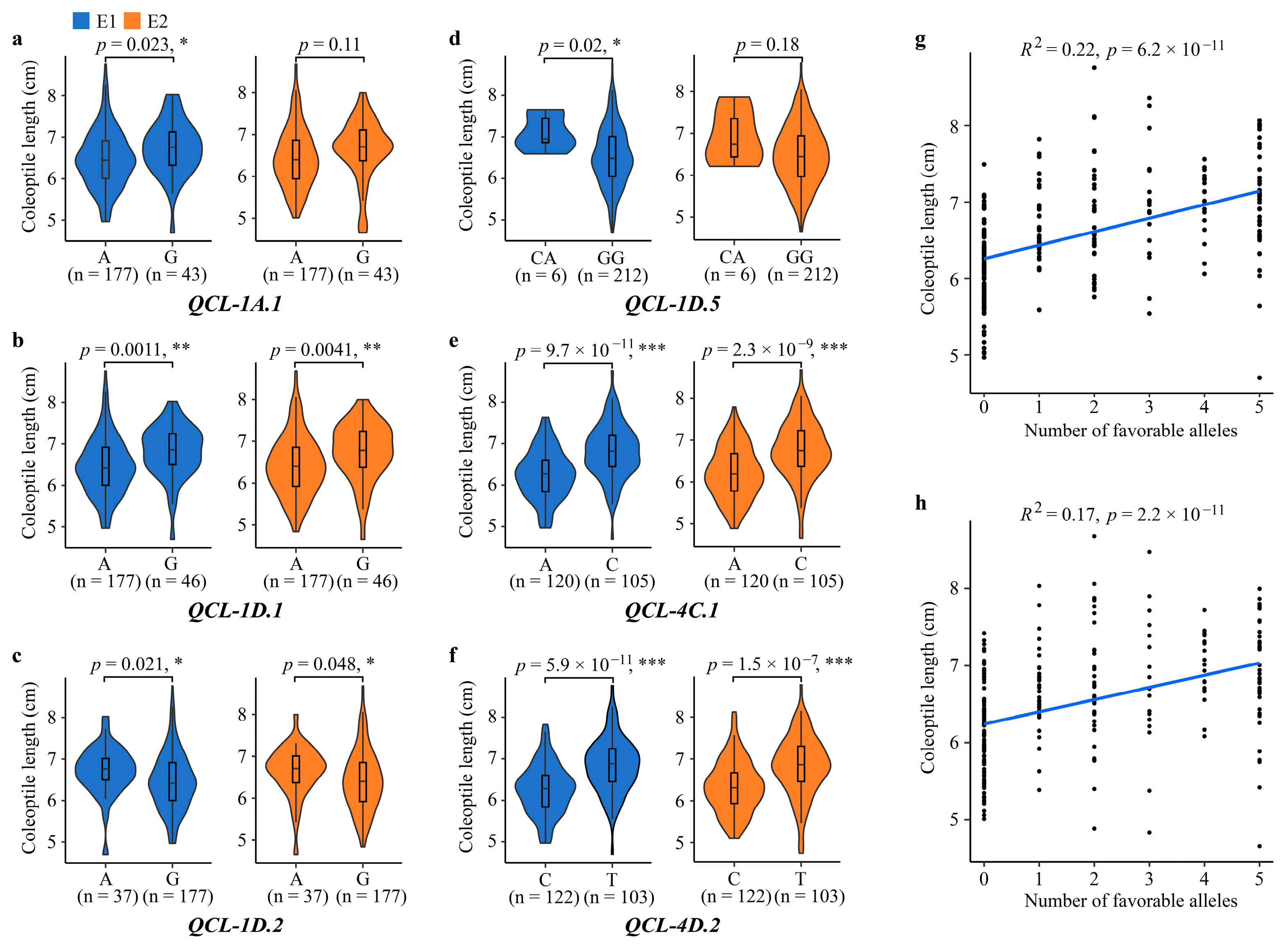
3.4. Effects of Favorable Allele on Coleoptile Length
3.5. Identification of Putative Candidate Genes
4. Discussion
4.1. Coleoptile Length Variation in Oat
4.2. Genomic Regions Associated with Coleoptile Length
4.3. Putative Candidate Genes for Coleoptile Length
4.4. Limitations of This Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, B.-L.; Zheng, Z.; Ren, C. Chapter 6—Oat. In Crop Physiology Case Histories for Major Crops; Sadras, V.O., Calderini, D.F., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 222–248. [Google Scholar]
- Esvelt Klos, K.; Huang, Y.-F.; Bekele, W.A.; Obert, D.E.; Babiker, E.; Beattie, A.D.; Bjørnstad, Å.; Bonman, J.M.; Carson, M.L.; Chao, S.; et al. Population genomics related to adaptation in elite oat germplasm. Plant Genome 2016, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Barsila, S.R.; Resources, N. The fodder oat (Avena sativa) mixed legume forages farming: Nutritional and ecological benefits. J. Agric. Nat. Resour. 2018, 1, 206–222. [Google Scholar] [CrossRef]
- Achleitner, A.; Tinker, N.A.; Zechner, E.; Buerstmayr, H. Genetic diversity among oat varieties of worldwide origin and associations of AFLP markers with quantitative traits. Theor. Appl. Genet. 2008, 117, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Radford, B.J.; Key, A.J. Temperature affects germination, mesocotyl length and coleoptile length of oats genotypes. Aust. J. Agric. Res. 1993, 44, 677–688. [Google Scholar] [CrossRef]
- Brown, P.R.; Singleton, G.R.; Tann, C.R.; Mock, I. Increasing sowing depth to reduce mouse damage to winter crops. Crop Prot. 2003, 22, 653–660. [Google Scholar] [CrossRef]
- O’Sullivan, P.A.; Weiss, G.M.; Friesen, D. Tolerance of spring wheat (Triticum aestivum L.) to trifluralin deep-incorporated in the autumn or spring. Weed Res. 1985, 25, 275–280. [Google Scholar] [CrossRef]
- Schillinger, W.F.; Donaldson, E.; Allan, R.E.; Jones, S.S. Winter wheat seedling emergence from deep sowing depths. Agron. J. 1998, 90, 582–586. [Google Scholar] [CrossRef]
- Kirby, E.J.M. Effect of sowing depth on seedling emergence, growth and development in barley and wheat. Field Crops Res. 1993, 35, 101–111. [Google Scholar] [CrossRef]
- Yang, W.; Xu, D.; Li, S.; Tang, X.; Pan, S.; Chen, X.; Mo, Z. Emergence and seedling establishment of rice varieties at different sowing depths. J. Plant Growth Regul. 2022, 41, 1672–1686. [Google Scholar] [CrossRef]
- Ma, J.; Lin, Y.; Tang, S.; Duan, S.; Wang, Q.; Wu, F.; Li, C.; Jiang, X.; Zhou, K.; Liu, Y. A Genome-wide association study of coleoptile length in different Chinese wheat landraces. Front. Plant Sci. 2020, 11, 677. [Google Scholar] [CrossRef]
- Rebetzke, G.J.; Richards, R.A.; Fettell, N.A.; Long, M.; Condon, A.G.; Forrester, R.I.; Botwright, T.L. Genotypic increases in coleoptile length improves stand establishment, vigour and grain yield of deep-sown wheat. Field Crops Res. 2007, 100, 10–23. [Google Scholar] [CrossRef]
- Bovill, W.D.; Hyles, J.; Zwart, A.B.; Ford, B.A.; Perera, G.; Phongkham, T.; Brooks, B.J.; Rebetzke, G.J.; Hayden, M.J.; Hunt, J.R.; et al. Increase in coleoptile length and establishment by Lcol-A1, a genetic locus with major effect in wheat. BMC Plant Biol. 2019, 19, 332. [Google Scholar] [CrossRef] [PubMed]
- Nghi, K.N.; Tondelli, A.; Valè, G.; Tagliani, A.; Marè, C.; Perata, P.; Pucciariello, C. Dissection of coleoptile elongation in japonica rice under submergence through integrated genome-wide association mapping and transcriptional analyses. Plant Cell Environ. 2019, 42, 1832–1846. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Bai, G.; Carver, B.F.; Elliott, N.C.; Bennett, R.S.; Wu, Y.; Hunger, R.; Bonman, J.M.; Xu, X. Genome-wide association study reveals genetic architecture of coleoptile length in wheat. Theor. Appl. Genet. 2017, 130, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Zhang, S.; Liu, Y.; Wang, J.; Wu, B.; Zhao, J.; Qiao, L.; Zheng, X.; Wang, J.; Zheng, J. Genome-wide association study of coleoptile length with Shanxi wheat. Front. Plant Sci. 2022, 13, 1016551. [Google Scholar] [CrossRef] [PubMed]
- Paynter, B.H.; Clarke, G.P.Y. Coleoptile length of barley (Hordeum vulgare L.) cultivars. Genet. Resour. Crop Evol. 2010, 57, 395–403. [Google Scholar] [CrossRef]
- Luo, H.; Hill, C.B.; Zhou, G.; Zhang, X.-Q.; Li, C. Genome-wide association mapping reveals novel genes associated with coleoptile length in a worldwide collection of barley. BMC Plant Biol. 2020, 20, 346. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, M.L. Coleoptile length and emergence in varieties of barley, oats, and wheat. Can. J. Plant Sci. 1968, 48, 357–361. [Google Scholar] [CrossRef]
- Koçak, B.; Kilinc, F.; Bardak, A.; Güngör, H.; Dokuyucu, T.; Akkaya, A.; Dumlupinar, Z. Association mapping of germination and some early seedling stage traits of a Turkish origin oat collection. Turk. J. Field Crops. 2022, 27, 41–50. [Google Scholar] [CrossRef]
- Murphy, K.; Balow, K.; Lyon, S.R.; Jones, S.S. Response to selection, combining ability and heritability of coleoptile length in winter wheat. Euphytica 2008, 164, 709–718. [Google Scholar] [CrossRef]
- Redoña, E.D.; Mackill, D.J. Mapping quantitative trait loci for seedling vigor in rice using RFLPs. Theor. Appl. Genet. 1996, 92, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-K.; Tung, C.-W. Genetic Mapping of anaerobic germination-associated QTLs controlling coleoptile elongation in rice. Rice 2015, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Li, S.; Zhang, C.; Qiang, Y.; Li, Y. Combination of quantitative trait locus (QTL) mapping and transcriptome analysis reveals submerged germination QTLs and candidate genes controlling coleoptile length in rice. Food Energy Secur. 2022, 11, e354. [Google Scholar] [CrossRef]
- Blackburn, A.; Sidhu, G.; Schillinger, W.F.; Skinner, D.; Gill, K. QTL mapping using GBS and SSR genotyping reveals genomic regions controlling wheat coleoptile length and seedling emergence. Euphytica 2021, 217, 45. [Google Scholar] [CrossRef]
- Singh, K.; Shukla, S.; Kadam, S.; Semwal, V.K.; Singh, N.K.; Khanna-Chopra, R. Genomic regions and underlying candidate genes associated with coleoptile length under deep sowing conditions in a wheat RIL population. J. Plant Biochem. Biot. 2015, 24, 324–330. [Google Scholar] [CrossRef]
- Rebetzke, G.J.; Appels, R.; Morrison, A.D.; Richards, R.A.; McDonald, G.; Ellis, M.H.; Spielmeyer, W.; Bonnett, D.G. Quantitative trait loci on chromosome 4B for coleoptile length and early vigour in wheat (Triticum aestivum L.). Aust. J. Agric. Res. 2001, 52, 1221–1234. [Google Scholar] [CrossRef]
- Rebetzke, G.J.; Ellis, M.H.; Bonnett, D.G.; Richards, R.A. Molecular mapping of genes for coleoptile growth in bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2007, 114, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Rebetzke, G.J.; Verbyla, A.P.; Verbyla, K.L.; Morell, M.K.; Cavanagh, C.R. Use of a large multiparent wheat mapping population in genomic dissection of coleoptile and seedling growth. Plant Biotechnol. J. 2014, 12, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Noda, M.; Sakurai, K.; Watanabe, A.; Akagi, H.; Sato, K.; Takeda, K. QTLs in barley controlling seedling elongation of deep-sown seeds. Euphytica 2008, 164, 761–768. [Google Scholar] [CrossRef]
- Takahashi, H.; Sato, K.; Takeda, K. Mapping genes for deep-seeding tolerance in barley. Euphytica 2001, 122, 37–43. [Google Scholar] [CrossRef]
- Tinker, N.A.; Wight, C.P.; Bekele, W.A.; Yan, W.; Jellen, E.N.; Renhuldt, N.T.; Sirijovski, N.; Lux, T.; Spannagl, M.; Mascher, M. Genome analysis in Avena sativa reveals hidden breeding barriers and opportunities for oat improvement. Commun. Biol. 2022, 5, 474. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Deng, D.; Zhou, P.; Peng, Y.; Dong, X.; Li, S.; Zhang, Y.; Man, Q.; Lv, Z.; Chen, T.; et al. Dissecting the genetic basis of grain weight and size in common oat by genome-wide association study. J. Cereal Sci. 2023, 114, 103811. [Google Scholar] [CrossRef]
- Peng, Y.; Yan, H.; Guo, L.; Deng, C.; Wang, C.; Wang, Y.; Kang, L.; Zhou, P.; Yu, K.; Dong, X.; et al. Reference genome assemblies reveal the origin and evolution of allohexaploid oat. Nat. Genet. 2022, 54, 1248–1258. [Google Scholar] [CrossRef]
- Kamal, N.; Tsardakas Renhuldt, N.; Bentzer, J.; Gundlach, H.; Haberer, G.; Juhász, A.; Lux, T.; Bose, U.; Tye-Din, J.A.; Lang, D.; et al. The mosaic oat genome gives insights into a uniquely healthy cereal crop. Nature 2022, 606, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhang, H.; Zhou, P.; Ren, C.; Peng, Y. Genome-wide association mapping of QTL underlying groat protein content of a diverse panel of oat accessions. Int. J. Mol. Sci. 2023, 24, 5581. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Kuehl, R.O.; Ray, I.M.; Hui, R.; Soleri, D. Evaluation of simple methods for estimating broad-sense heritability in stands of randomly planted genotypes. Crop Sci. 1998, 38, 1125–1129. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinf. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, J.S.; Singh, D.; Gill, H.S.; Brar, N.K.; Qiu, Y.; Halder, J.; Al Tameemi, R.; Turnipseed, B.; Sehgal, S.K. Genome-wide association study uncovers novel genomic regions associated with coleoptile length in hard winter wheat. Front. Plant Sci. 2020, 10, 1345. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Chaban, C.; Waller, F.; Furuya, M.; Nick, P. Auxin responsiveness of a novel cytochrome P450 in rice coleoptiles. Plant Physiol. 2003, 133, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Nghi, K.N.; Tagliani, A.; Mariotti, L.; Weits, D.A.; Perata, P.; Pucciariello, C. Auxin is required for the long coleoptile trait in japonica rice under submergence. New Phytol. 2021, 229, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Canales, F.J.; Montilla-Bascón, G.; Bekele, W.A.; Howarth, C.J.; Langdon, T.; Rispail, N.; Tinker, N.A.; Prats, E. Population genomics of Mediterranean oat (A. sativa) reveals high genetic diversity and three loci for heading date. Theor. Appl. Genet. 2021, 134, 2063–2077. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Richards, D.E.; Hartley, N.M.; Murphy, G.P.; Devos, K.M.; Flintham, J.E.; Beales, J.; Fish, L.J.; Worland, A.J.; Pelica, F.; et al. ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 1999, 400, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.H.; Rebetzke, G.J.; Chandler, P.; Bonnett, D.; Spielmeyer, W.; Richards, R.A. The effect of different height reducing genes on the early growth of wheat. Funct. Plant Biol. 2004, 31, 583–589. [Google Scholar] [CrossRef]
- Cheng, J.; Hill, C.; Han, Y.; He, T.; Ye, X.; Shabala, S.; Guo, G.; Zhou, M.; Wang, K.; Li, C. New semi-dwarfing alleles with increased coleoptile length by gene editing of gibberellin 3-oxidase 1 using CRISPR-Cas9 in barley (Hordeum vulgare L.). Plant Biotechnol. J. 2023, 21, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Paque, S.; Weijers, D. Q&A: Auxin: The plant molecule that influences almost anything. BMC Biol. 2016, 14, 67. [Google Scholar]
- Blakeslee, J.J.; Peer, W.A.; Murphy, A.S. Auxin transport. Curr. Opin. Plant Biol. 2005, 8, 494–500. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | df a | Sum of Squares | Mean Squares | F-Values | Significance b |
|---|---|---|---|---|---|
| Genotype | 242 | 717.3 | 2.96 | 38.91 | *** |
| Experiment | 1 | 0.9 | 0.90 | 11.77 | *** |
| G × E | 242 | 32.0 | 0.13 | 1.74 | *** |
| Residuals | 972 | 74.0 | 0.08 |
| Variables | Range (cm) | Mean (cm) | Standard Deviation (cm) | CV (%) | H2 |
|---|---|---|---|---|---|
| E1 | 4.70–8.75 | 6.57 | 0.71 | 10.77 | |
| E2 | 4.66–8.68 | 6.52 | 0.73 | 11.13 | |
| Total | 4.66–8.75 | 6.55 | 0.75 | 11.45 | 0.86 |
| QTL | Marker | Chromosome | Position (Mb) | Allele * | E1 | E2 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| −Log10 (p) | R2 (%) | Allelic Effects (%) | −Log10 (p) | R2 (%) | Allelic Effects (%) | |||||
| QCL-1A.1 | S1A_295026052 | 1A | 295.03 | A/G | 3.72 | 8.11 | 4.05 | 3.82 | 8.35 | 3.09 |
| QCL-1D.1 | S1D_38632063 | 1D | 38.63 | A/G | 3.54 | 7.64 | 5.57 | 3.16 | 6.83 | 5.27 |
| QCL-1D.2 | S1D_49143792 | 1D | 49.14 | G/A | 3.37 | 7.77 | 4.03 | 3.51 | 8.05 | 3.54 |
| QCL-1D.5 | S1D_180046647 | 1D | 180.05 | G/C | 3.36 | 7.09 | 6.05 | 3.05 | 6.44 | 4.30 |
| S1D_180046702 | 1D | 180.05 | G/A | 3.43 | 7.42 | 8.84 | 3.09 | 6.43 | 6.54 | |
| QCL-4C.1 | S4C_36128238 | 4C | 36.13 | A/C | 4.38 | 10.07 | 9.05 | 3.25 | 7.30 | 8.79 |
| QCL-4D.2 | S4D_350378493 | 4D | 350.38 | C/T | 3.53 | 8.07 | 9.26 | 3.31 | 7.69 | 7.86 |
| QTL | Gene_ID | Distance (bp) to Significant SNP # | Description | Literature |
|---|---|---|---|---|
| QCL-1A.1 | A.satnudSFS1A01G005205 | −1,936,883 | Cytochrome P450 85A1-like | Chaban et al. [43] |
| A.satnudSFS1A01G005149 | 1,517,948 | Cytochrome P450 71A1 | Chaban et al. [43] | |
| A.satnudSFS1A01G005147 | 1,682,924 | Cytochrome P450 71A1 | Chaban et al. [43] | |
| QCL-4C.1 | A.satnudSFS4C01G001030 | 1,742,832 | Cytochrome P450 72A15 | Chaban et al. [43] |
| QCL-4D.2 | A.satnudSFS4D01G002954 | 1,583,082 | Auxin transporter-like protein 1 | Nghi et al. [44] |
| QCL-4D.2 | A.satnudSFS4D01G002963 | 1,233,083 | Cytochrome P450 CBH32594.1 | Chaban et al. [43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, P.; Liu, Y.; Yang, M.; Yan, H. Genome-Wide Association Study Uncovers Genomic Regions Associated with Coleoptile Length in a Worldwide Collection of Oat. Genes 2024, 15, 411. https://doi.org/10.3390/genes15040411
Zhou P, Liu Y, Yang M, Yan H. Genome-Wide Association Study Uncovers Genomic Regions Associated with Coleoptile Length in a Worldwide Collection of Oat. Genes. 2024; 15(4):411. https://doi.org/10.3390/genes15040411
Chicago/Turabian StyleZhou, Pingping, Yuankun Liu, Mengxian Yang, and Honghai Yan. 2024. "Genome-Wide Association Study Uncovers Genomic Regions Associated with Coleoptile Length in a Worldwide Collection of Oat" Genes 15, no. 4: 411. https://doi.org/10.3390/genes15040411