
Impact of 12-SNP and 6-SNP Polygenic Scores on Predisposition to High LDL-Cholesterol Levels in Patients with Familial Hypercholesterolemia
 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Biochemical Data
2.3. Genetic Screening
2.4. Statistical Analysis
3. Results
3.1. Identification of Monogenic Causes of Hypercholesterolemia
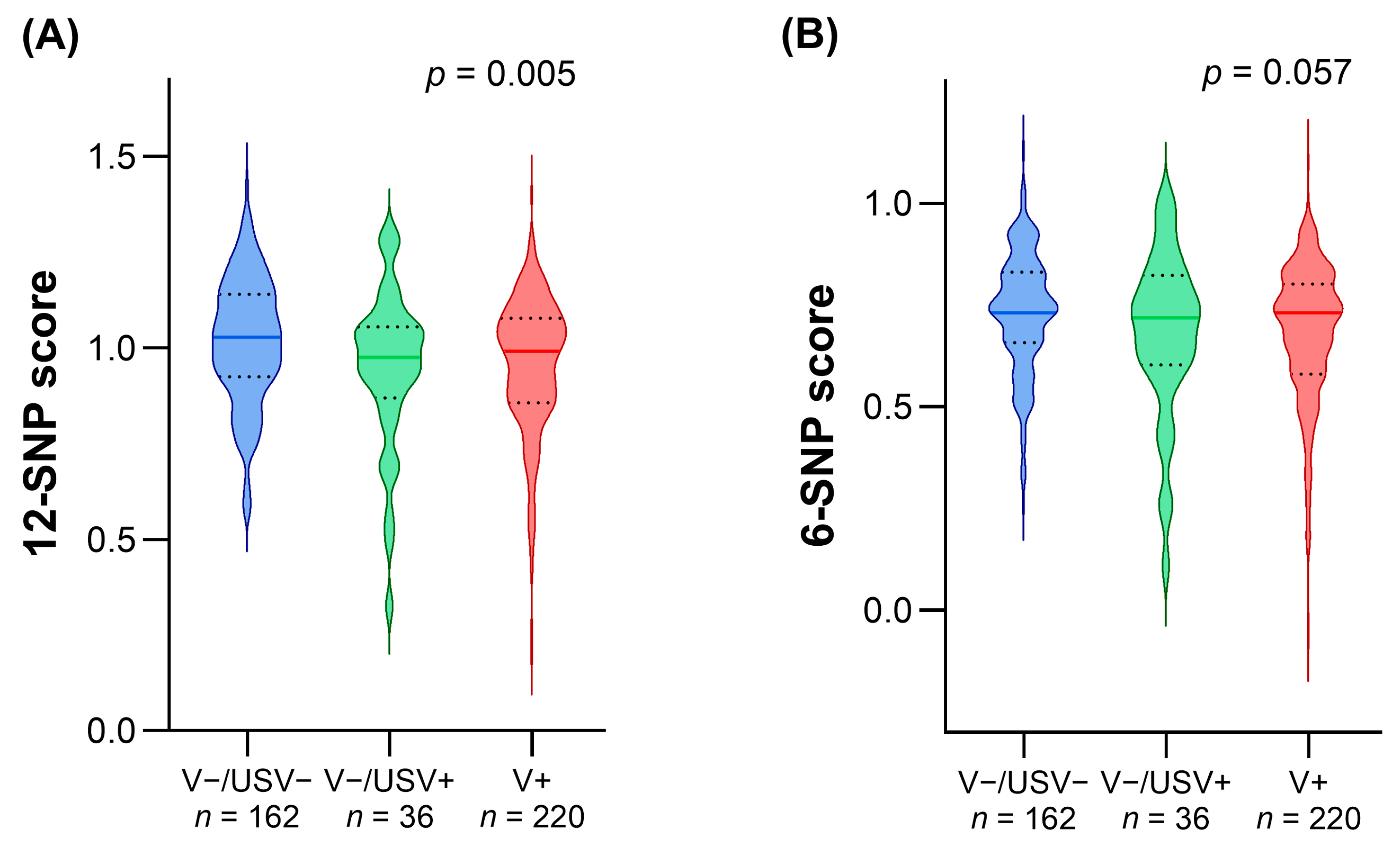
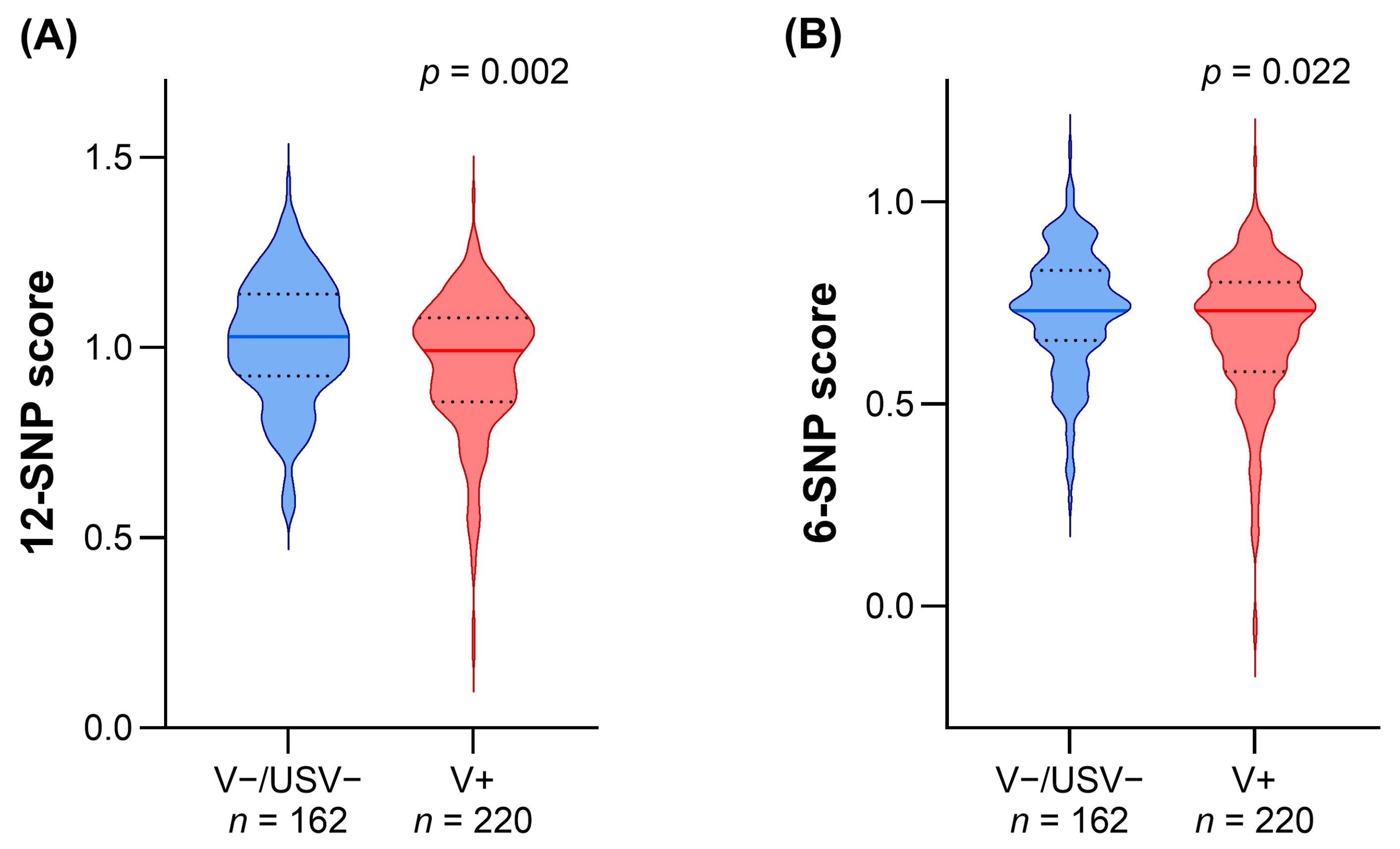
3.2. Comparison of LDL-c Polygenic Scores in Patients with Different Genetic Statuses
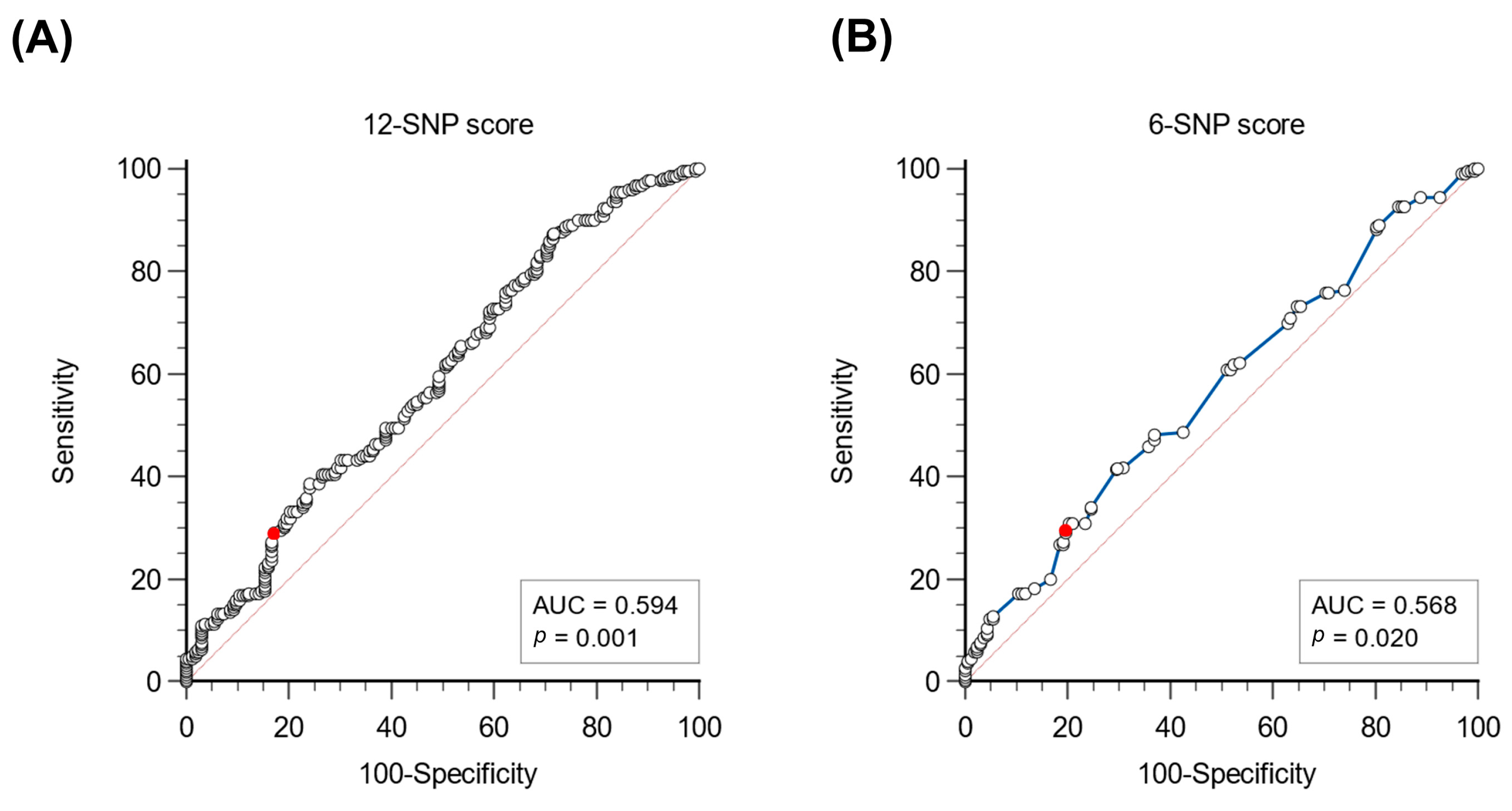
3.3. Evaluation of a Cut-Off to Identify Patients with a Polygenic Basis of Hypercholesterolemia
3.4. Association of Polygenic Scores with Lipid Traits/LDL-Cholesterol Levels
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Watts, G.F.; Gidding, S.S.; Hegele, R.A.; Raal, F.J.; Sturm, A.C.; Jones, L.K.; Sarkies, M.N.; Al-Rasadi, K.; Blom, D.J.; Daccord, M.; et al. International Atherosclerosis Society Guidance for Implementing Best Practice in the Care of Familial Hypercholesterolaemia. Nat. Rev. Cardiol. 2023, 20, 845–869. [Google Scholar] [CrossRef]
- Akioyamen, L.E.; Genest, J.; Shan, S.D.; Reel, R.L.; Albaum, J.M.; Chu, A.; Tu, J.V. Estimating the Prevalence of Heterozygous Familial Hypercholesterolaemia: A Systematic Review and Meta-Analysis. BMJ Open 2017, 7, e016461. [Google Scholar] [CrossRef]
- Tromp, T.R.; Hartgers, M.L.; Hovingh, G.K.; Vallejo-Vaz, A.J.; Ray, K.K.; Soran, H.; Freiberger, T.; Bertolini, S.; Harada-Shiba, M.; Blom, D.J.; et al. Worldwide Experience of Homozygous Familial Hypercholesterolaemia: Retrospective Cohort Study. Lancet 2022, 399, 719–728. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; Giacobbe, C.; Buonaiuto, A.; Calcaterra, I.; Palma, D.; Maione, G.; Iannuzzo, G.; Di Minno, M.N.D.; Rubba, P.; Fortunato, G. A Real-World Experience of Clinical, Biochemical and Genetic Assessment of Patients with Homozygous Familial Hypercholesterolemia. J. Clin. Med. 2020, 9, 219. [Google Scholar] [CrossRef]
- Alves, A.C.; Alonso, R.; Diaz-Diaz, J.L.; Medeiros, A.M.; Jannes, C.E.; Merchan, A.; Vasques-Cardenas, N.A.; Cuevas, A.; Chacra, A.P.; Krieger, J.E.; et al. Phenotypical, Clinical, and Molecular Aspects of Adults and Children With Homozygous Familial Hypercholesterolemia in Iberoamerica. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2508–2515. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; Fortunato, G. Genetic Heterogeneity of Familial Hypercholesterolemia: Repercussions for Molecular Diagnosis. Int. J. Mol. Sci. 2023, 24, 3224. [Google Scholar] [CrossRef]
- Abifadel, M.; Boileau, C. Genetic and Molecular Architecture of Familial Hypercholesterolemia. J. Intern. Med. 2023, 293, 144–165. [Google Scholar] [CrossRef]
- Tada, H.; Okada, H.; Nomura, A.; Yashiro, S.; Nohara, A.; Ishigaki, Y.; Takamura, M.; Kawashiri, M.-A. Rare and Deleterious Mutations in ABCG5/ABCG8 Genes Contribute to Mimicking and Worsening of Familial Hypercholesterolemia Phenotype. Circ. J. 2019, 83, 1917–1924. [Google Scholar] [CrossRef]
- Tada, H.; Nohara, A.; Kawashiri, M.-A. Monogenic, Polygenic, and Oligogenic Familial Hypercholesterolemia. Curr. Opin. Lipidol. 2019, 30, 300–306. [Google Scholar] [CrossRef]
- Khera, A.V.; Won, H.-H.; Peloso, G.M.; Lawson, K.S.; Bartz, T.M.; Deng, X.; van Leeuwen, E.M.; Natarajan, P.; Emdin, C.A.; Bick, A.G.; et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 2016, 67, 2578–2589. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; Giacobbe, C.; Palma, D.; Iannuzzo, G.; Gentile, M.; Calcaterra, I.; Guardamagna, O.; Auricchio, R.; Di Minno, M.N.D.; Fortunato, G. Genetic Spectrum of Familial Hypercholesterolemia and Correlations with Clinical Expression: Implications for Diagnosis Improvement. Clin. Genet. 2021, 100, 529–541. [Google Scholar] [CrossRef]
- Romano, M.; Di Taranto, M.D.; Mirabelli, P.; D’Agostino, M.N.; Iannuzzi, A.; Marotta, G.; Gentile, M.; Raia, M.; Di Noto, R.; Del Vecchio, L.; et al. An Improved Method on Stimulated T-Lymphocytes to Functionally Characterize Novel and Known LDLR Mutations. J. Lipid Res. 2011, 52, 2095–2100. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in Atherosclerotic Cardiovascular Disease and Aortic Stenosis: A European Atherosclerosis Society Consensus Statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Olmastroni, E.; Gazzotti, M.; Averna, M.; Arca, M.; Tarugi, P.; Calandra, S.; Bertolini, S.; Catapano, A.L.; Casula, M.; LIPIGEN Study Group. Lipoprotein(a) Genotype Influences the Clinical Diagnosis of Familial Hypercholesterolemia. J. Am. Heart Assoc. 2023, 12, e029223. [Google Scholar] [CrossRef]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, Clinical and Population Relevance of 95 Loci for Blood Lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Talmud, P.J.; Shah, S.; Whittall, R.; Futema, M.; Howard, P.; Cooper, J.A.; Harrison, S.C.; Li, K.; Drenos, F.; Karpe, F.; et al. Use of Low-Density Lipoprotein Cholesterol Gene Score to Distinguish Patients with Polygenic and Monogenic Familial Hypercholesterolaemia: A Case-Control Study. Lancet 2013, 381, 1293–1301. [Google Scholar] [CrossRef]
- Futema, M.; Shah, S.; Cooper, J.A.; Li, K.; Whittall, R.A.; Sharifi, M.; Goldberg, O.; Drogari, E.; Mollaki, V.; Wiegman, A.; et al. Refinement of Variant Selection for the LDL Cholesterol Genetic Risk Score in the Diagnosis of the Polygenic Form of Clinical Familial Hypercholesterolemia and Replication in Samples from 6 Countries. Clin. Chem. 2015, 61, 231–238. [Google Scholar] [CrossRef]
- Wang, J.; Dron, J.S.; Ban, M.R.; Robinson, J.F.; McIntyre, A.D.; Alazzam, M.; Zhao, P.J.; Dilliott, A.A.; Cao, H.; Huff, M.W.; et al. Polygenic Versus Monogenic Causes of Hypercholesterolemia Ascertained Clinically. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2439–2445. [Google Scholar] [CrossRef]
- Tada, H.; Yeo, K.K.; Li, J.-J.; Tan, K.; Ako, J.; Krittayaphong, R.; San Tan, R.; Aylward, P.E.; Lam, C.S.P.; Baek, S.H.; et al. Polygenic Risk Scores for Atherosclerotic Cardiovascular Disease in the Asia-Pacific Region. JACC Asia 2021, 1, 294–302. [Google Scholar] [CrossRef]
- Gratton, J.; Finan, C.; Hingorani, A.D.; Humphries, S.E.; Futema, M. LDL-C Concentrations and the 12-SNP LDL-C Score for Polygenic Hypercholesterolaemia in Self-Reported South Asian, Black and Caribbean Participants of the UK Biobank. Front. Genet. 2022, 13, 845498. [Google Scholar] [CrossRef]
- Wiegman, A.; Gidding, S.S.; Watts, G.F.; Chapman, M.J.; Ginsberg, H.N.; Cuchel, M.; Ose, L.; Averna, M.; Boileau, C.; Borén, J.; et al. Familial Hypercholesterolaemia in Children and Adolescents: Gaining Decades of Life by Optimizing Detection and Treatment. Eur. Heart J. 2015, 36, 2425–2437. [Google Scholar] [CrossRef] [PubMed]
- Di Taranto, M.D.; de Falco, R.; Guardamagna, O.; Massini, G.; Giacobbe, C.; Auricchio, R.; Malamisura, B.; Proto, M.; Palma, D.; Greco, L.; et al. Lipid Profile and Genetic Status in a Familial Hypercholesterolemia Pediatric Population: Exploring the LDL/HDL Ratio. Clin. Chem. Lab. Med. 2019, 57, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Langsted, A.; Kamstrup, P.R.; Benn, M.; Tybjærg-Hansen, A.; Nordestgaard, B.G. High Lipoprotein(a) as a Possible Cause of Clinical Familial Hypercholesterolaemia: A Prospective Cohort Study. Lancet Diabetes Endocrinol. 2016, 4, 577–587. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Chora, J.R.; Medeiros, A.M.; Alves, A.C.; Bourbon, M. Analysis of Publicly Available LDLR, APOB, and PCSK9 Variants Associated with Familial Hypercholesterolemia: Application of ACMG Guidelines and Implications for Familial Hypercholesterolemia Diagnosis. Genet. Med. 2018, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Chora, J.R.; Iacocca, M.A.; Tichý, L.; Wand, H.; Kurtz, C.L.; Zimmermann, H.; Leon, A.; Williams, M.; Humphries, S.E.; Hooper, A.J.; et al. The Clinical Genome Resource (ClinGen) Familial Hypercholesterolemia Variant Curation Expert Panel Consensus Guidelines for LDLR Variant Classification. Genet. Med. 2022, 24, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Rubba, P.; Gentile, M.; Marotta, G.; Iannuzzi, A.; Sodano, M.; De Simone, B.; Jossa, F.; Iannuzzo, G.; Giacobbe, C.; Di Taranto, M.D.; et al. Causative Mutations and Premature Cardiovascular Disease in Patients with Heterozygous Familial Hypercholesterolaemia. Eur. J. Prev. Cardiol. 2017, 24, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Saadatagah, S.; Alhalabi, L.; Farwati, M.; Zordok, M.; Bhat, A.; Smith, C.Y.; Wood-Wentz, C.M.; Bailey, K.R.; Kullo, I.J. The Burden of Severe Hypercholesterolemia and Familial Hypercholesterolemia in a Population-Based Setting in the US. Am. J. Prev. Cardiol. 2022, 12, 100393. [Google Scholar] [CrossRef] [PubMed]
- D’Erasmo, L.; Minicocci, I.; Di Costanzo, A.; Pigna, G.; Commodari, D.; Ceci, F.; Montali, A.; Brancato, F.; Stanca, I.; Nicolucci, A.; et al. Clinical Implications of Monogenic Versus Polygenic Hypercholesterolemia: Long-Term Response to Treatment, Coronary Atherosclerosis Burden, and Cardiovascular Events. J. Am. Heart Assoc. 2021, 10, e018932. [Google Scholar] [CrossRef]
- Olmastroni, E.; Gazzotti, M.; Arca, M.; Averna, M.; Pirillo, A.; Catapano, A.L.; Casula, M.; LIPIGEN Study Group. Twelve Variants Polygenic Score for Low-Density Lipoprotein Cholesterol Distribution in a Large Cohort of Patients With Clinically Diagnosed Familial Hypercholesterolemia With or Without Causative Mutations. J. Am. Heart Assoc. 2022, 11, e023668. [Google Scholar] [CrossRef]
- Schwaninger, G.; Forer, L.; Ebenbichler, C.; Dieplinger, H.; Kronenberg, F.; Zschocke, J.; Witsch-Baumgartner, M. Filling the Gap: Genetic Risk Assessment in Hypercholesterolemia Using LDL-C and LPA Genetic Scores. Clin. Genet. 2023, 104, 334–343. [Google Scholar] [CrossRef]
- Sjouke, B.; Tanck, M.W.T.; Fouchier, S.W.; Defesche, J.C.; Hutten, B.A.; Wiegman, A.; Kastelein, J.J.P.; Hovingh, G.K. Children with Hypercholesterolemia of Unknown Cause: Value of Genetic Risk Scores. J. Clin. Lipidol. 2016, 10, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Ripatti, P.; Rämö, J.T.; Mars, N.J.; Fu, Y.; Lin, J.; Söderlund, S.; Benner, C.; Surakka, I.; Kiiskinen, T.; Havulinna, A.S.; et al. Polygenic Hyperlipidemias and Coronary Artery Disease Risk. Circ. Genom. Precis. Med. 2020, 13, e002725. [Google Scholar] [CrossRef] [PubMed]
- Sinnott-Armstrong, N.; Tanigawa, Y.; Amar, D.; Mars, N.; Benner, C.; Aguirre, M.; Venkataraman, G.R.; Wainberg, M.; Ollila, H.M.; Kiiskinen, T.; et al. Genetics of 35 Blood and Urine Biomarkers in the UK Biobank. Nat. Genet. 2021, 53, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.E.; Clarke, S.L.; Wu, K.-H.H.; Kanoni, S.; Zajac, G.J.M.; Ramdas, S.; Surakka, I.; Ntalla, I.; Vedantam, S.; Winkler, T.W.; et al. The Power of Genetic Diversity in Genome-Wide Association Studies of Lipids. Nature 2021, 600, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Camastra, F.; Di Taranto, M.D.; Staiano, A. Statistical and Computational Methods for Genetic Diseases: An Overview. Comput. Math. Methods Med. 2015, 2015, 954598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhan, J.; Jin, J.; Zhang, J.; Lu, W.; Zhao, R.; Ahearn, T.U.; Yu, Z.; O’Connell, J.; Jiang, Y.; et al. A New Method for Multiancestry Polygenic Prediction Improves Performance across Diverse Populations. Nat. Genet. 2023, 55, 1757–1768. [Google Scholar] [CrossRef]
- Mariano, C.; Alves, A.C.; Medeiros, A.M.; Chora, J.R.; Antunes, M.; Futema, M.; Humphries, S.E.; Bourbon, M. The Familial Hypercholesterolaemia Phenotype: Monogenic Familial Hypercholesterolaemia, Polygenic Hypercholesterolaemia and Other Causes. Clin. Genet. 2020, 97, 457–466. [Google Scholar] [CrossRef]
- Cuchel, M.; Bruckert, E.; Ginsberg, H.N.; Raal, F.J.; Santos, R.D.; Hegele, R.A.; Kuivenhoven, J.A.; Nordestgaard, B.G.; Descamps, O.S.; Steinhagen-Thiessen, E.; et al. Homozygous Familial Hypercholesterolaemia: New Insights and Guidance for Clinicians to Improve Detection and Clinical Management. A Position Paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 2014, 35, 2146–2157. [Google Scholar] [CrossRef]
- Gazzotti, M.; Casula, M.; Bertolini, S.; Capra, M.E.; Olmastroni, E.; Catapano, A.L.; Pederiva, C.; LIPIGEN Paediatric Group. The Role of Registers in Increasing Knowledge and Improving Management of Children and Adolescents Affected by Familial Hypercholesterolemia: The LIPIGEN Pediatric Group. Front. Genet. 2022, 13, 912510. [Google Scholar] [CrossRef]
- Awan, Z.A.; Rashidi, O.M.; Al-Shehri, B.A.; Jamil, K.; Elango, R.; Al-Aama, J.Y.; Hegele, R.A.; Banaganapalli, B.; Shaik, N.A. Saudi Familial Hypercholesterolemia Patients With Rare LDLR Stop Gain Variant Showed Variable Clinical Phenotype and Resistance to Multiple Drug Regimen. Front. Med. 2021, 8, 694668. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, A.M.; Alves, A.C.; Miranda, B.; Chora, J.R.; Bourbon, M.; Investigators of the Portuguese FH Study. Unraveling the Genetic Background of Individuals with a Clinical Familial Hypercholesterolemia Phenotype. J. Lipid Res. 2024, 65, 100490. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.R.; Tada, M.T.; Oliveira, T.G.M.; Jannes, C.E.; Bensenor, I.; Lotufo, P.A.; Santos, R.D.; Krieger, J.E.; Pereira, A.C. Polygenic Risk Score for Hypercholesterolemia in a Brazilian Familial Hypercholesterolemia Cohort. Atheroscler. Plus 2022, 49, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zamora, A.; Ramos, R.; Comas-Cufi, M.; García-Gil, M.; Martí-Lluch, R.; Plana, N.; Alves-Cabratosa, L.; Ponjoan, A.; Rodríguez-Borjabad, C.; Ibarretxe, D.; et al. Women with Familial Hypercholesterolemia Phenotype Are Undertreated and Poorly Controlled Compared to Men. Sci. Rep. 2023, 13, 1492. [Google Scholar] [CrossRef] [PubMed]
- Staiano, A.; di Taranto, M.D.; Bloise, E.; D’Agostino, M.N.; D’Angelo, A.; Marotta, G.; Gentile, M.; Jossa, F.; Iannuzzi, A.; Rubba, P.; et al. Investigation of Single Nucleotide Polymorphisms Associated to Familial Combined Hyperlipidemia with Random Forests. In Neural Nets and Surroundings; Smart Innovation, Systems and Technologies; Springer: Berlin/Heidelberg, Germany, 2013; Volume 19, pp. 169–178. [Google Scholar] [CrossRef]
- Civeira, F.; Jarauta, E.; Cenarro, A.; García-Otín, A.L.; Tejedor, D.; Zambón, D.; Mallen, M.; Ros, E.; Pocoví, M. Frequency of Low-Density Lipoprotein Receptor Gene Mutations in Patients with a Clinical Diagnosis of Familial Combined Hyperlipidemia in a Clinical Setting. J. Am. Coll. Cardiol. 2008, 52, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Dron, J.S. The Clinical Utility of Polygenic Risk Scores for Combined Hyperlipidemia. Curr. Opin. Lipidol. 2023, 34, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Ripatti, P.; Rämö, J.T.; Söderlund, S.; Surakka, I.; Matikainen, N.; Pirinen, M.; Pajukanta, P.; Sarin, A.-P.; Service, S.K.; Laurila, P.-P.; et al. The Contribution of GWAS Loci in Familial Dyslipidemias. PLoS Genet. 2016, 12, e1006078. [Google Scholar] [CrossRef]
- Di Minno, M.N.D.; Gentile, M.; Di Minno, A.; Iannuzzo, G.; Calcaterra, I.; Buonaiuto, A.; Di Taranto, M.D.; Giacobbe, C.; Fortunato, G.; Rubba, P.O.F. Changes in Carotid Stiffness in Patients with Familial Hypercholesterolemia Treated with Evolocumab®: A Prospective Cohort Study. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Trinder, M.; Li, X.; DeCastro, M.L.; Cermakova, L.; Sadananda, S.; Jackson, L.M.; Azizi, H.; Mancini, G.B.J.; Francis, G.A.; Frohlich, J.; et al. Risk of Premature Atherosclerotic Disease in Patients With Monogenic Versus Polygenic Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2019, 74, 512–522. [Google Scholar] [CrossRef]
- Trinder, M.; Francis, G.A.; Brunham, L.R. Association of Monogenic vs Polygenic Hypercholesterolemia With Risk of Atherosclerotic Cardiovascular Disease. JAMA Cardiol. 2020, 5, 390–399. [Google Scholar] [CrossRef]
- Pfisterer, S.G.; Brock, I.; Kanerva, K.; Hlushchenko, I.; Paavolainen, L.; Ripatti, P.; Islam, M.M.; Kyttälä, A.; Di Taranto, M.D.; Scotto di Frega, A.; et al. Multiparametric Platform for Profiling Lipid Trafficking in Human Leukocytes. Cell Rep. Methods 2022, 2, 100166. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J.P.; Ginsberg, H.N.; Langslet, G.; Hovingh, G.K.; Ceska, R.; Dufour, R.; Blom, D.; Civeira, F.; Krempf, M.; Lorenzato, C.; et al. ODYSSEY FH I and FH II: 78 Week Results with Alirocumab Treatment in 735 Patients with Heterozygous Familial Hypercholesterolaemia. Eur. Heart J. 2015, 36, 2996–3003. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, R.P.; Pedersen, T.R.; Park, J.-G.; De Ferrari, G.M.; Gaciong, Z.A.; Ceska, R.; Toth, K.; Gouni-Berthold, I.; Lopez-Miranda, J.; Schiele, F.; et al. Clinical Efficacy and Safety of Achieving Very Low LDL-Cholesterol Concentrations with the PCSK9 Inhibitor Evolocumab: A Prespecified Secondary Analysis of the FOURIER Trial. Lancet 2017, 390, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Total FH Patients n = 418 | Adults n = 285 | Children n = 133 | Significance 1 | FH/V+ Patients n = 220 | FH/V− Patients n = 198 | Significance 2 | |
|---|---|---|---|---|---|---|---|
| Age (years) | 31 (14–54) | 47 (30–58) | 11 (9–13) | p < 0.001 | 20 (12–44) | 45 (21–58) | p < 0.001 |
| Sex n males (%) | 188.0 (45%) | 120.0 (42.1%) | 68.0 (51.1%) | ns | 103.0 (46.8%) | 85.0 (42.9%) | ns |
| Pediatric patients n (%) | 133.0 (31.8%) | - | - | - | 91.0 (41.4%) | 42.0 (21.2%) | p < 0.001 |
| LDL-cholesterol (mg/dL) | 209 (176–251) | 213 (181–260) | 198 (155–232) | p < 0.001 | 215 (183–263) | 198 (172–234) | p = 0.004 |
| Total cholesterol (mg/dL) | 295 (253–345) | 303 (262–367) | 271 (230–312) | p < 0.001 | 299 (253–350) | 293 (253–332) | ns |
| HDL-cholesterol (mg/dL) | 52 (44–62) | 51 (43–62) | 54 (47–64) | p = 0.041 | 52 (45–61) | 53 (44–67) | ns |
| Non HDL-cholesterol (mg/dL) | 241 (200–292) | 249 (211–313) | 214 (177–257) | p < 0.001 | 245 (203–298) | 236 (198–284) | ns |
| Triglycerides (mg/dL) | 102 (75–155) | 114 (84–166) | 82 (65–110) | p < 0.001 | 91 (67–142) | 114 (86–166) | p < 0.001 |
| LDL/HDL ratio | 4.0 (3.1–5.2) | 4.3 (3.4–5.5) | 3.5 (2.8–4.5) | p < 0.001 | 4.2 (3.2–5.4) | 3.9 (2.9–4.9) | p = 0.012 |
| Presence of pathogenic variants n (%) | 220.0 (52.6%) | 129.0 (45.3%) | 91.0 (68.4%) | p < 0.001 | - | - | - |
| FH/V−/USV− n = 162 | FH/V+ n = 220 | Significance | |
|---|---|---|---|
| 12-SNP score − | 32 (19.8%) | 70 (31.2%) | p = 0.010 |
| 12-SNP score + | 130 (80.2%) | 150 (68.2%) | |
| 6-SNP score − | 32 (19.8%) | 65 (29.5%) | p = 0.033 |
| 6-SNP score + | 130 (80.2%) | 155 (70.5%) |
| Total FH Patients n = 382 | Mann-Whitney 1 | ANCOVA 2 | FH/V−/USV− n = 162 | Mann-Whitney 1 | ANCOVA 2 | FH/V+ n = 220 | Mann-Whitney 1 | ANCOVA 2 | |
|---|---|---|---|---|---|---|---|---|---|
| 12-SNP score − | 210 (165–239) | ns | p = 0.027 | 204 (169–231) | ns | ns | 212 (162–240) | p = 0.017 | p = 0.006 |
| 12-SNP score + | 209 (177–261) | 197 (173–236) | 223 (187–279) | ||||||
| 6-SNP score − | 213 (163–241) | ns | ns | 204 (166–231) | ns | ns | 215 (159–246) | p = 0.044 | p = 0.048 |
| 6-SNP score + | 209 (178–259) | 197 (173–236) | 217 (187–279) |
| Total FH Patients n = 382 | FH/V−/USV− n = 162 | FH/V+ n = 220 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12-SNP Score | 6-SNP Score | 12-SNP Score | 6-SNP Score | 12-SNP Score | 6-SNP Score | |||||||
| β-Coefficient | p-Value | β-Coefficient | p-Value | β-Coefficient | p-Value | β-Coefficient | p-Value | β-Coefficient | p-Value | β-Coefficient | p-Value | |
| LDL-cholesterol (mg/dL) | 0.144 | p = 0.005 | 0.134 | p = 0.009 | 0.104 | ns | 0.032 | ns | 0.212 | p = 0.002 | 0.227 | p < 0.001 |
| Total cholesterol (mg/dL) | 0.120 | p = 0.020 | 0.111 | p = 0.031 | 0.072 | ns | 0.021 | ns | 0.169 | p = 0.012 | 0.183 | p = 0.007 |
| HDL-cholesterol (mg/dL) | −0.003 | ns | −0.003 | ns | 0.055 | ns | 0.092 | ns | −0.069 | ns | −0.089 | ns |
| Non-HDL cholesterol (mg/dL) | 0.127 | p = 0.014 | 0.118 | p = 0.022 | 0.068 | ns | 0.008 | ns | 0.186 | p = 0.006 | 0.203 | p = 0.002 |
| Triglycerides (mg/dL) | 0.045 | ns | 0.026 | ns | −0.015 | ns | −0.050 | ns | 0.026 | ns | 0.030 | ns |
| LDL/HDL ratio | 0.089 | ns | 0.085 | ns | 0.041 | ns | −0.029 | ns | 0.157 | p = 0.021 | 0.186 | p = 0.006 |
| Total Population n = 382 | FH/V+ Patients n = 220 | FH/V−/USV− Patients n = 160 | Adults n = 257 | Children n = 125 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Independent Variables | β | Significance | β | Significance | β | Significance | β | Significance | β | Significance |
| Age | 0.274 | p < 0.001 | 0.151 | p = 0.025 | 0.415 | p < 0.001 | 0.218 | p = 0.001 | −0.052 | ns |
| Sex | 0.017 | ns | 0.044 | ns | −0.010 | ns | 0.023 | ns | 0.043 | ns |
| Presence of pathogenic variants | 0.240 | p < 0.001 | - | - | - | - | 0.175 | p = 0.007 | 0.407 | p < 0.001 |
| 12-SNP score | 0.169 | p = 0.001 | 0.199 | p = 0.003 | 0.154 | p = 0.037 | 0.149 | p = 0.016 | 0.242 | p = 0.005 |
| Total Population n = 382 | FH/V+ Patients n = 220 | FH/V−/USV− Patients n = 160 | Adults n = 257 | Children n = 125 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Independent Variables | β | Significance | β | Significance | β | Significance | β | Significance | β | Significance |
| Age | 0.273 | p < 0.001 | 0.149 | p = 0.027 | 0.405 | p < 0.001 | 0.213 | p = 0.001 | −0.043 | ns |
| Sex | 0.017 | ns | 0.038 | ns | 0.003 | ns | 0.027 | ns | 0.029 | ns |
| Presence of pathogenic variants | 0.233 | p < 0.001 | - | - | - | - | 0.166 | p = 0.009 | 0.400 | p < 0.001 |
| 6-SNP score | 0.152 | p = 0.002 | 0.211 | p = 0.002 | 0.083 | ns | 0.146 | p = 0.017 | 0.201 | p = 0.021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardiero, G.; Ferrandino, M.; Calcaterra, I.L.; Iannuzzo, G.; Di Minno, M.N.D.; Buganza, R.; Guardamagna, O.; Auricchio, R.; Di Taranto, M.D.; Fortunato, G. Impact of 12-SNP and 6-SNP Polygenic Scores on Predisposition to High LDL-Cholesterol Levels in Patients with Familial Hypercholesterolemia. Genes 2024, 15, 462. https://doi.org/10.3390/genes15040462
Cardiero G, Ferrandino M, Calcaterra IL, Iannuzzo G, Di Minno MND, Buganza R, Guardamagna O, Auricchio R, Di Taranto MD, Fortunato G. Impact of 12-SNP and 6-SNP Polygenic Scores on Predisposition to High LDL-Cholesterol Levels in Patients with Familial Hypercholesterolemia. Genes. 2024; 15(4):462. https://doi.org/10.3390/genes15040462
Chicago/Turabian StyleCardiero, Giovanna, Martina Ferrandino, Ilenia Lorenza Calcaterra, Gabriella Iannuzzo, Matteo Nicola Dario Di Minno, Raffaele Buganza, Ornella Guardamagna, Renata Auricchio, Maria Donata Di Taranto, and Giuliana Fortunato. 2024. "Impact of 12-SNP and 6-SNP Polygenic Scores on Predisposition to High LDL-Cholesterol Levels in Patients with Familial Hypercholesterolemia" Genes 15, no. 4: 462. https://doi.org/10.3390/genes15040462