
Genetic Modifiers of Sickle Cell Anemia Phenotype in a Cohort of Angolan Children

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Assessment of Hematological and Biochemical Parameters
2.2. Sample Characterization
2.3. Targeted Sequencing
2.4. Variant Calling Quality Control and Annotation
2.5. Variant Annotation and Mutation Prioritization
2.6. Network and Enrichment Analysis
2.7. Principal Component Analysis (PCA)
2.8. Distribution of Minor Allele Frequency and Gene-Specific in SNP Frequencies
2.9. Identity by Descent (IBD) and Functional Genomics
2.10. HbF Association Testing
2.11. Meta-Analysis of Angolan HbF and Other African Ancestry HbF
2.12. Rare-Variant Association and Burden Tests
2.13. Estimating Functional Heritability from GWAS Dataset
3. Results
3.1. Participant Characteristics and Targeted Variant Discovery
3.2. In Silico Mutational Burden of Genes in Participants
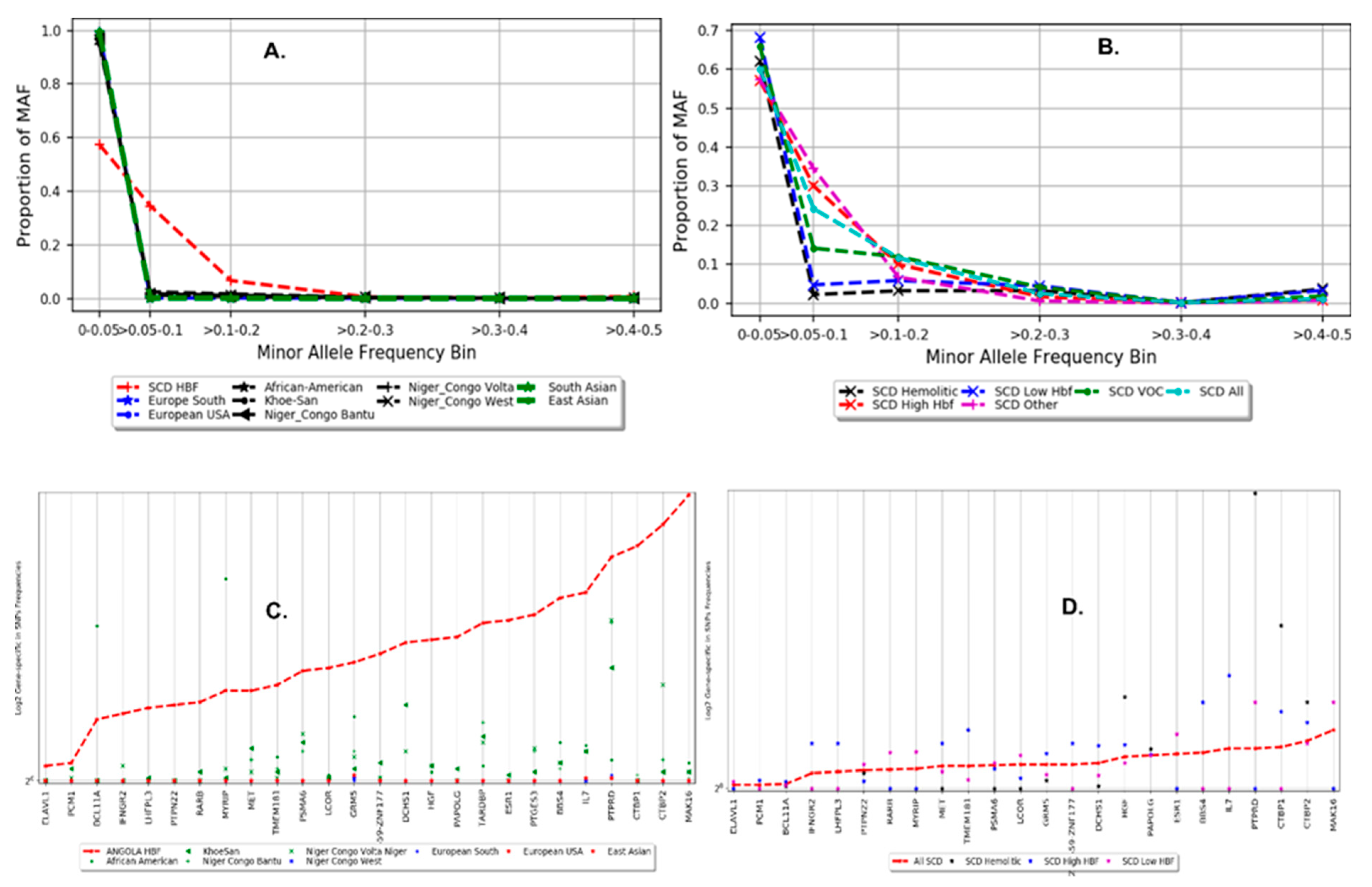
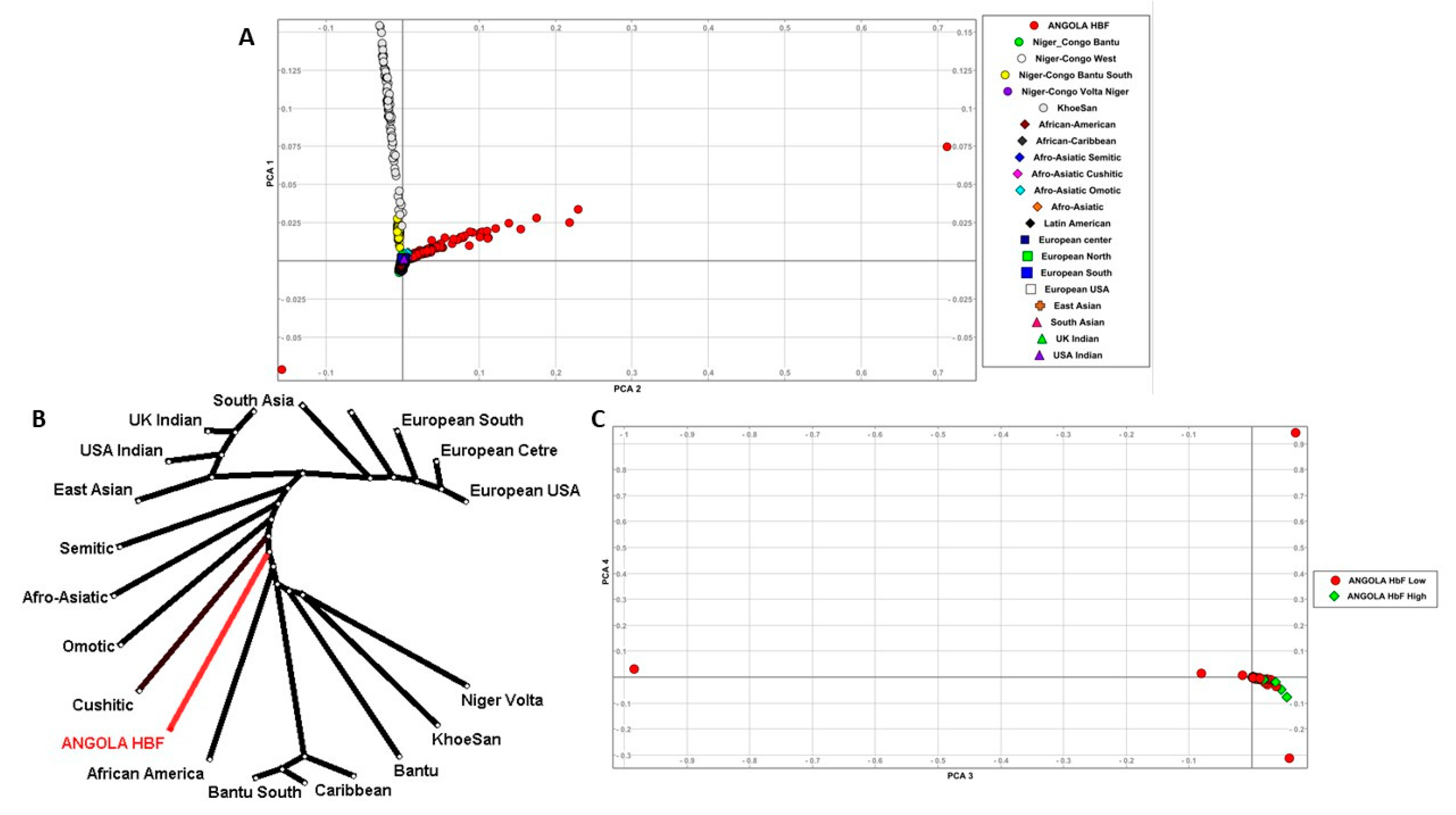
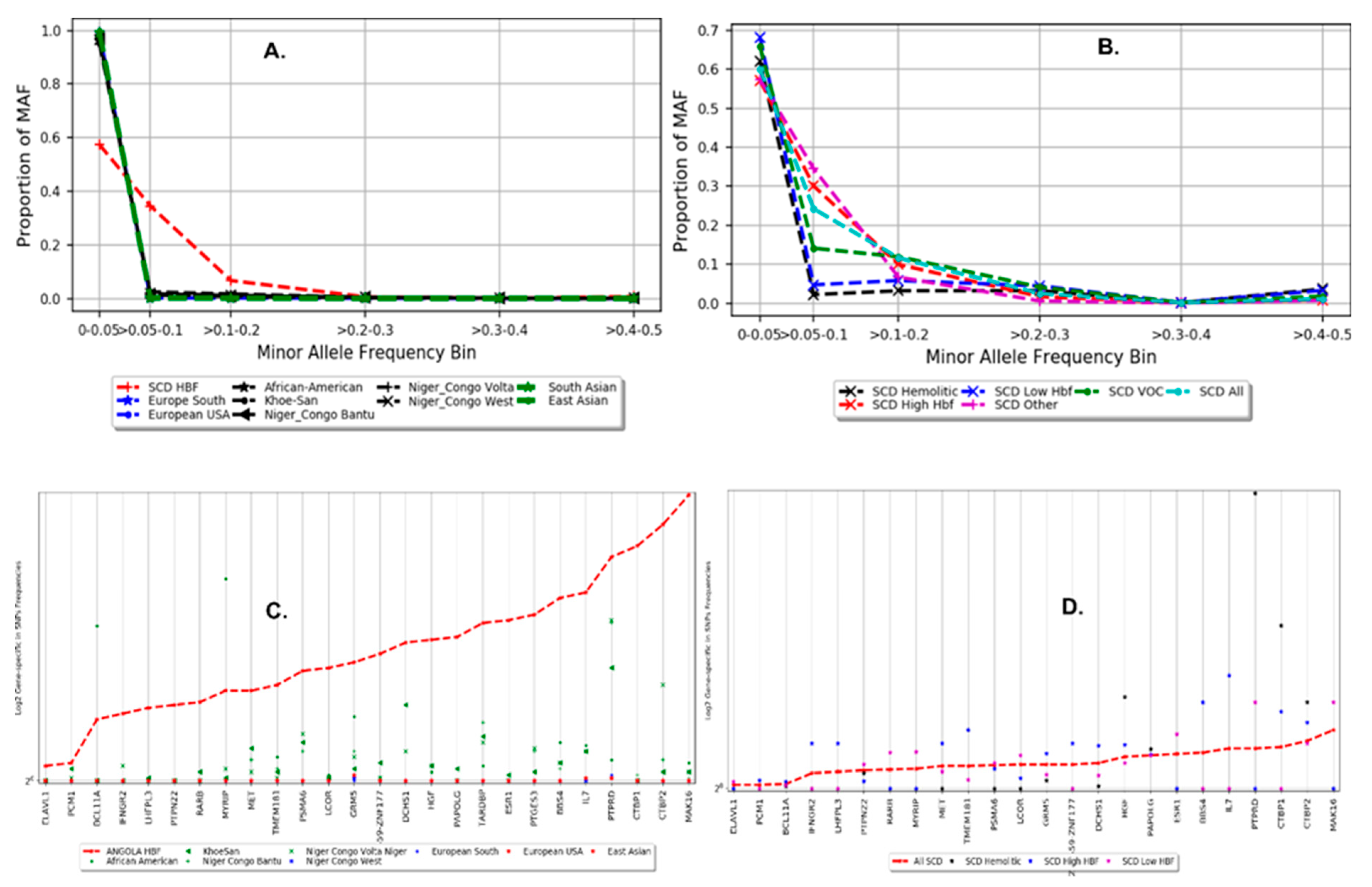
3.3. Population Structure and Distribution of Gene-Specific in SNP Frequencies
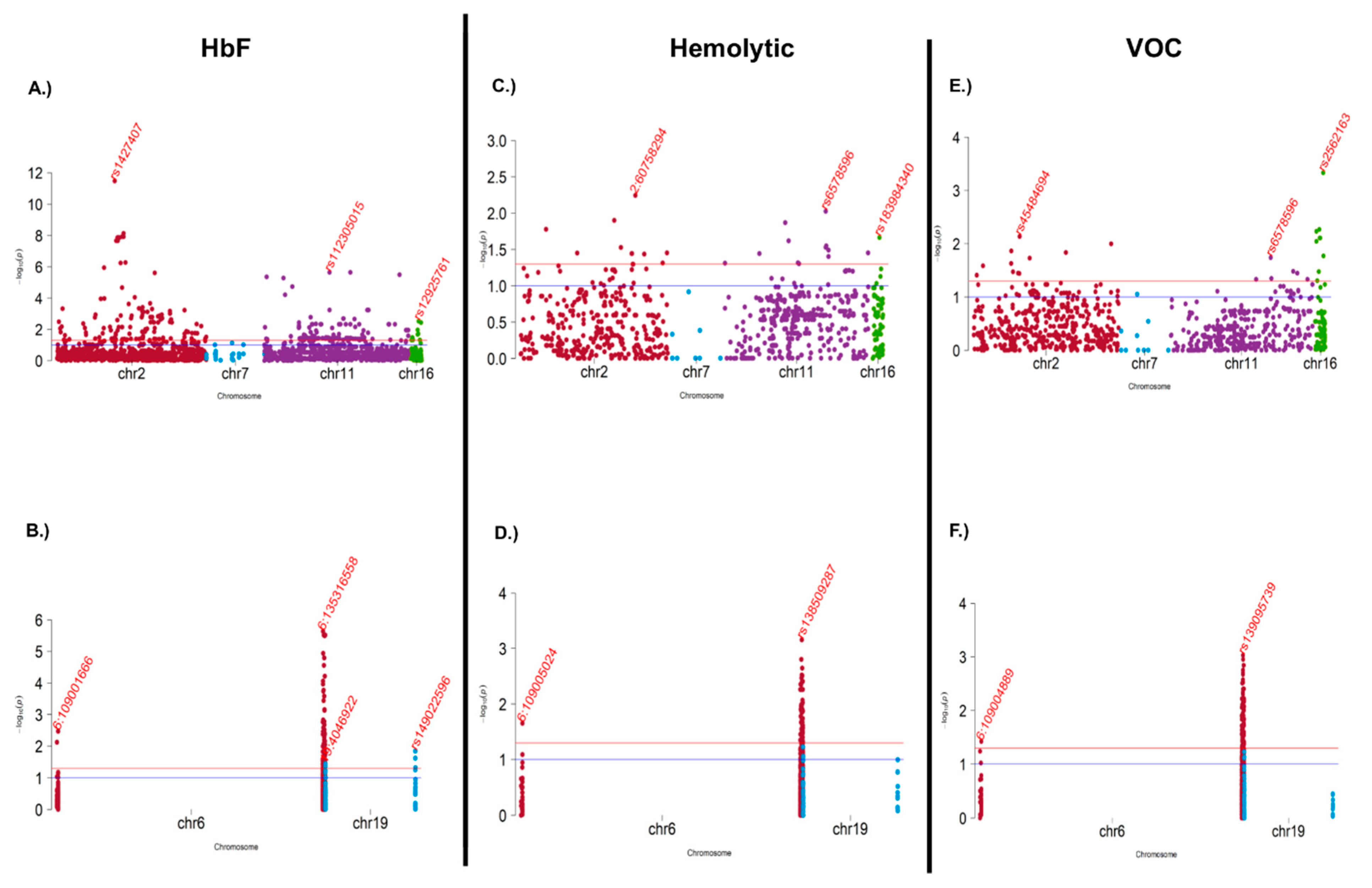
3.4. Association and Meta-Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Prim. 2018, 4, 18010. [Google Scholar] [CrossRef]
- Chang, A.K.; Ginter Summarell, C.C.; Birdie, P.T.; Sheehan, V.A. Genetic modifiers of severity in sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Hay, S.I.; Gupta, S.; Weatherall, D.J.; Williams, T.N. Global Burden of Sickle Cell Anaemia in Children under Five, 2010-2050: Modelling Based on Demographics, Excess Mortality, and Interventions. PLoS Med. 2013, 10, e1001484. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Sebastiani, P. Genetic modifiers of sickle cell disease. Am. J. Hematol. 2012, 87, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Lettre, G.; Bauer, D.E. Fetal haemoglobin in sickle-cell disease: From genetic epidemiology to new therapeutic strategies. Lancet 2016, 387, 2554–2564. [Google Scholar] [CrossRef] [PubMed]
- Sales, R.R.; Belisário, A.R.; Faria, G.; Mendes, F.; Luizon, M.R.; Viana, M.B. Functional polymorphisms of BCL11A and HBS1L-MYB genes affect both fetal hemoglobin level and clinical outcomes in a cohort of children with sickle cell anemia. Ann. Hematol. 2020, 99, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Brouard, J.S.; Schenkel, F.; Marete, A.; Bissonnette, N. The GATK joint genotyping workflow is appropriate for calling variants in RNA-seq experiments. J. Anim. Sci. Biotechnol. 2019, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Gézsi, A.; Bolgár, B.; Marx, P.; Sarkozy, P.; Szalai, C.; Antal, P. VariantMetaCaller: Automated fusion of variant calling pipelines for quantitative, precision-based filtering. BMC Genom. 2015, 16, 875. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2; John Wiley & Sons: Hoboken, NJ, USA, 2013; Volume 2. [Google Scholar]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 2013, 29, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef]
- Kim, S.; Jhong, J.H.; Lee, J.; Koo, J.Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017, 10, 2. [Google Scholar]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Ionita-Laza, I.; Mccallum, K.; Buxbaum, J. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016, 48, 214–220. [Google Scholar] [CrossRef]
- Lu, Q.; Hu, Y.; Sun, J.; Cheng, Y.; Cheung, K.H.; Zhao, H. A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data. Sci. Rep. 2015, 5, 10576. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Schmidt, S.; Peshkin, L.; Ramensky, V.; Gerasimova, A.; Bork, P.; Kondrashov, A.; Sunyaev, S. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Doerks, T.; Copley, R.R.; Schultz, J.; Ponting, C.P.; Bork, P. Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 2002, 12, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.; Guttman, M.; Clamp, M.; Zody, M.C.; Friedman, N.; Xie, X. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 2009, 25, i54–i62. [Google Scholar] [CrossRef]
- Zuberi, K.; Franz, M.; Rodriguez, H.; Montojo, J.; Lopes, C.T.; Bader, G.D.; Morris, Q. GeneMANIA prediction server 2013 update. Nucleic Acids Res. 2013, 41, W115–W122. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Loh, P.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; Reshef, A.; Finucane, H.K.; Schoenherr, S.; Forer, L.; Mccarthy, S.; Abecasis, G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Gurdasani, D.; Carstensen, T.; Tekola-Ayele, F.; Pagani, L.; Tachmazidou, I.; Hatzikotoulas, K.; Karthikeyan, S.; Iles, L.; Pollard, M.O.; Choudhury, A.; et al. The African Genome Variation Project shapes medical genetics in Africa. Nature 2015, 517, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Gudykunst, W.B.; Schmidt, K.L. Language and Ethnic identity: An overview and prologue. J. Lang. Soc. Psychol. 1987, 6, 157–170. [Google Scholar] [CrossRef]
- Michalopoulos, S. The Origins of Ethnolinguistic Diversit. Am. Econ. Rev. 2012, 102, 1508–1539. [Google Scholar] [CrossRef] [PubMed]
- Wonkam, A.; Chimusa, E.R.; Mnika, K.; Pule, G.D.; Ngo Bitoungui, V.J.; Mulder, N.; Shriner, D.; Rotimi, C.N.; Adeyemo, A. Genetic modifiers of long-term survival in sickle cell anemia. Clin. Transl. Med. 2020, 10, e152. [Google Scholar] [CrossRef]
- Chimusa, E.R.; Beighton, P.; Kumuthini, J.; Ramesar, R.S. Detecting genetic modifiers of spondyloepimetaphyseal dysplasia with joint laxity in the Caucasian Afrikaner community. Hum. Mol. Genet. 2019, 28, 1053–1063. [Google Scholar] [CrossRef]
- Seidman, D.N.; Shenoy, S.A.; Kim, M.; Babu, R.; Woods, I.G.; Dyer, T.D.; Lehman, D.M.; Curran, J.E.; Duggirala, R.; Blangero, J.; et al. Rapid, Phase-free Detection of Long Identity-by-Descent Segments Enables Effective Relationship Classification. Am. J. Hum. Genet. 2020, 106, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef]
- Mtatiro, S.N.; Singh, T.; Rooks, H.; Mgaya, J.; Mariki, H.; Soka, D.; Mmbando, B.; Msaki, E.; Kolder, I.; Thein, S.L.; et al. Genome wide association study of fetal hemoglobin in sickle cell Anemia in Tanzania. PLoS ONE 2014, 9, e111464. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Pertsemlidis, A.; Ding, L.H.; Story, M.D.; Steinberg, M.H.; Sebastiani, P.; Hoppe, C.; Ballas, S.K.; Pace, B.S. Original Research: A case-control genome-wide association study identifies genetic modifiers of fetal hemoglobin in sickle cell disease. Exp. Biol. Med. 2016, 241, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Eskin, E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 2011, 88, 586–598. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Xu, B.; Lee, S.; Ionita-Laza, I. Unified Sequence-Based Association Tests Allowing for Multiple Functional Annotations and Meta-analysis of Noncoding Variation in Metabochip Data. Am. J. Hum. Genet. 2017, 101, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Togninalli, M.; Roqueiro, D.; Borgwardt, K.M. Accurate and adaptive imputation of summary statistics in mixed-ethnicity cohorts. Bioinformatics 2018, 34, i687–i696. [Google Scholar] [CrossRef] [PubMed]
- Pasaniuc, B.; Zaitlen, N.; Shi, H.; Bhatia, G.; Gusev, A.; Pickrell, J.; Hirschhorn, J.; Strachan, D.P.; Patterson, N.; Price, A.L. Fast and accurate imputation of summary statistics enhances evidence of functional enrichment. Bioinformatics 2014, 30, 2906–2914. [Google Scholar] [CrossRef] [PubMed]
- Gazal, S.; Marquez-Luna, C.; Finucane, H.K.; Price, A.L. Reconciling S-LDSC and LDAK functional enrichment estimates. Nat. Genet. 2019, 51, 1202–1204. [Google Scholar] [CrossRef] [PubMed]
- Speed, D.; Kaphle, A.; Balding, D.J. SNP-based heritability and selection analyses: Improved models and new results. BioEssays 2022, 44, e2100170. [Google Scholar] [CrossRef] [PubMed]
- Siva, N. 1000 Genomes project. Nat. Biotechnol. 2008, 26, 256–257. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Benz, E.J.; Adewoye, A.H.; Ebert, B.L. Pathobiology of the human erythrocyte and its hemoglobins. In Hematology: Basic Principles and Practice; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 447–457. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | HbF Groups | ||||||
|---|---|---|---|---|---|---|---|
| Hemolytic (n = 21) | Vaso-Oclusive (n = 138) | Less Severe (n = 33) | p-Value * (Kruskal–Wallis) | Low—HbF (n = 143) | High—HbF (n = 48) | p-Value (t-Test) | |
| Mean (SD) | Mean (SD) | Mean (SD) | Mean (SD) | Mean (SD) | |||
| Fetal Hemoglobin (%) | 5.11 (3.07) | 5.62 (3.94) | 6.14 (4.61) | 0.765 | 3.89 (1.96) | 10.92 (3.79) | - |
| Hemoglobin (g/dL) | 7.02 (1.01) | 7.38 (0.98) | 7.34 (0.89) | 0.188 | 7.17 (0.91) | 7.81 (1.01) | <0.001 |
| Reticulocyte (%) | 11.55 (3.2) | 9.94 (4.84) | 11.1 (4.54) | 0.022 | 10.43 (4.75) | 9.98 (4.44) | 0.999 |
| Erythrocytes (1012L) | 2.68 (0.45) | 3 (0.65) | 2.88 (0.56) | 0.059 | 2.9 (0.61) | 3.07 (0.66) | 0.999 |
| MCV (fL) | 80.1 (6.41) | 76.45 (9.21) | 77.61 (7.67) | 0.072 | 76.7 (8.83) | 78.02 (8.55) | 0.999 |
| MCH (pg) | 26.44 (2.37) | 25.12 (3.07) | 25.88 (2.81) | 0.098 | 25.2 (2.96) | 25.95 (3.01) | 0.999 |
| White blood cells (109L) | 13.48 (3.38) | 14.07 (5) | 14.12 (3.99) | 0.735 | 14.11 (4.87) | 13.77 (4.119 | 0.999 |
| Neutrophil (109L) | 6.15 (2.15) | 5.88 (2.34) | 5.91 (2.55) | 0.616 | 5.96 (2.31) | 5.82 (2.49) | 0.999 |
| Platelet (109L) | 382.32 (122.85) | 440.89 (180.74) | 448.97 (152.62) | 0.182 | 437.08 (180.36) | 432.8 (143.78) | 0.999 |
| Transfusions/year | 0.86 (0.91) | 0.34 (0.47) | 0.35 (0.32) | 0.009 | 0.46 (0.59) | 0.22 (0.31) | <0.001 |
| Hospitalizations/year | 0.9 (0.77) | 0.45 (0.47) | 0.37 (0.3) | 0.003 | 0.54 (0.54) | 0.31 (0.32) | 0.011 |
| Gene | CHR | Start Position | End Position | Band | Gene Biotype | p | Nbr Marker | Marker Tested | Marker Rare | Marker Common |
|---|---|---|---|---|---|---|---|---|---|---|
| LHFPL3 | chr7 | 104328603 | 104908561 | q22.2 | protein-coding | 1.65 × 10−5 | 40 | 31 | 31 | 9 |
| ZNF559-ZNF177 | chr19 | 9324174 | 9382617 | p13.2 | protein-coding | 0.00012 | 8 | 5 | 5 | 3 |
| TMEM181 | chr6 | 158536436 | 158635429 | q25.3 | protein-coding | 0.0033 | 40 | 29 | 29 | 11 |
| PAPOLG | chr2 | 60756253 | 60802086 | p16.1 | protein-coding | 0.0035 | 39 | 33 | 33 | 6 |
| CHR | BP | SNP | MAF | A1/A2 | Gene | Band | Func | β | SE | p |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 60718043 | rs1427407 | 0.22 | T/G | BCL11A | p16.1 | intronic | −3.11 | 0.49 | 1.29 × 10−9 |
| 2 | 60723096 | rs71327644 | 0.30 | C/CA | BCL11A | p16.1 | intronic | −2.55 | 0.46 | 7.39 × 10−8 |
| 2 | 60724087 | rs1896296 | 0.3 | G/T | BCL11A | p16.1 | intronic | −2.51 | 0.48 | 3.9 × 10−7 |
| 2 | 60724086 | rs1896295 | 0.29 | T/C | BCL11A | p16.1 | intronic | −2.45 | 0.48 | 7.36 × 10−7 |
| 2 | 60719970 | rs766432 | 0.3 | C/A | BCL11A | p16.1 | intronic | −2.43 | 0.48 | 9.38 × 10−7 |
| 2 | 60720589 | rs10195871 | 0.35 | A/G | BCL11A | p16.1 | intronic | −2.43 | 0.48 | 9.38 × 10−7 |
| 2 | 60720757 | rs10172646 | 0.32 | G/A | BCL11A | p16.1 | intronic | −2.43 | 0.48 | 9.38 × 10−7 |
| 2 | 60721347 | rs7557939 | 0.3 | G/A | BCL11A | p16.1 | intronic | −2.43 | 0.48 | 9.38 × 10−7 |
| 2 | 60720951 | rs4671393 | 0.3 | A/G | BCL11A | p16.1 | intronic | −2.43 | 0.48 | 9.38 × 10−7 |
| 2 | 60720318 | rs34211119 | 0.3 | gtt/gt | BCL11A | p16.1 | intronic | −2.43 | 0.48 | 9.38 × 10−7 |
| 2 | 60721311 | rs7584113 | 0.30 | A/G | BCL11A | p16.1 | intronic | −2.41 | 0.48 | 1.29 × 10−6 |
| 2 | 60720246 | rs11886868 | 0.3 | C/T | BCL11A | p16.1 | intronic | −2.37 | 0.48 | 1.72 × 10−6 |
| 2 | 60719074 | rs1896294 | 0.3 | C/T | BCL11A | p16.1 | intronic | −2.37 | 0.48 | 1.72 × 10−6 |
| 2 | 60725451 | rs7606173 | 0.42 | C/G | BCL11A | p16.1 | intronic | 1.81 | 0.41 | 2 × 10−5 |
| 2 | 60722040 | rs6706648 | 0.38 | T/C | BCL11A | p16.1 | intronic | 1.87 | 0.44 | 3.01 × 10−5 |
| 2 | 60710738 | rs11692396 | 0.25 | G/A | BCL11A | p16.1 | intronic | −1.98 | 0.5 | 9.03 × 10−5 |
| 11 | 51572589 | 1032:33:00 | 0.06 | T/G | OR4C46 | q11 | intergenic | −3.51 | 0.92 | 0.00019 |
| 2 | 60723108 | rs45606437 | 0.32 | A/AC | BCL11A | p16.1 | intronic | 1.61 | 0.43 | 0.00024 |
| 6 | 13542533 | rs1195623516 | 0.05 | T/C | GFOD1 | p23 | intergenic | −3.59 | 0.97 | 0.00029 |
| 6 | 13542532 | rs754078005 | 0.051 | A/G | GFOD1 | p23 | intergenic | −3.59 | 0.97 | 0.00029 |
| 2 | 92312693 | rs201391728 | 0.25 | G/T | ACTR3BP2 | p11.1 | intergenic | −1.97 | 0.55 | 0.00045 |
| 2 | 92307971 | rs201915260 | 0.18 | G/T | ACTR3BP2 | p11.1 | intergenic | 2.02 | 0.58 | 0.00057 |
| 7 | 100550995 | rs1394766104 | 0.088 | G/A | MUC3A | q22.1 | exonic | 2.64 | 0.78 | 0.00081 |
| 2 | 92312692 | rs200577446 | 0.22 | G/T | ACTR3BP2 | p11.1 | intergenic | −1.94 | 0.58 | 0.0009 |
| CHR | SNP | BP (hg19) | A1/A2 | p Values FE | OR ± STD FE | Pvalues R E | OR ± STD RE | p Values BE | P1 | P2 | P3 | M1 | M2 | M3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | rs147630502 | 60718043 | T/G | 5.0 × 10−29 | 0.2 ± 1.1 | 0.002 | 0.2 ± 1.7 | 4.2 × 10−30 | 1.2 × 10−9 | 2.2 × 10−24 | 0.17 | 1.0 | 1.0 | 0.2 |
| 2 | 2:60720951 | 60720951 | A/G | 3.9 × 10−23 | 0.2 ± 1.2 | 0.0005 | 0.2 ± 1.5 | 1.0 × 10−22 | 9.3 × 10−7 | 5.2 × 10−19 | 0.81 | 1.0 | 1.0 | 0.7 |
| 2 | 2:60719970 | 60719970 | C/A | 7.3 × 10−23 | 0.25 ± 1.1 | 0.0007 | 0.2 ± 1.5 | 1.5 × 10−22 | 9.38 × 10−7 | 8.1 × 10−19 | 0.64 | 1.0 | 1.0 | 0.7 |
| 2 | 2:60720757 | 60720757 | G/A | 1.3 × 10−19 | 0.3 ± 1.13 | 0.02 | 0.3 ± 1.7 | 2.8 × 10−22 | 9.38 × 10−7 | 9.04 × 10−19 | 0.79 | 1.00 | 1.0 | 0.7 |
| 2 | 2:60720589 | 60720589 | A/G | 2.2 × 10−19 | 0.3 ± 1.1 | 0.03 | 0.3 ± 1.7 | 2.2 × 10−22 | 9.38 × 10−7 | 7.7 × 10−19 | 0.95 | 0.9 | 1.0 | 0.6 |
| 2 | 2:60719074 | 60719074 | C/T | 2.0 × 10−18 | 0.3 ± 1.14 | 0.09 | 0.3 ± 1.9 | 2.2 × 10−23 | 1.71 × 10−6 | 6.1 × 10−20 | 0.31 | 1.0 | 1.0 | 0.0 |
| 2 | 2:60682447 | 60682447 | G/A | 1.2 × 10−12 | 2.6 ± 1.12 | 0.0003 | 2.2 ± 1.3 | 1.2 × 10−12 | 0.008 | 1.04 × 10−11 | 0.29 | 0.96 | 1.0 | 0.6 |
| 2 | 2:60755798 | 60755798 | T/C | 1.4 × 10−11 | 3.0 ± 1.17 | 1.4 × 10−11 | 2.9 ± 1.2 | 4.1 × 10−11 | 0.008 | 3.02 × 10−10 | 0.84 | 0.97 | 1.0 | 0.7 |
| 2 | 2:60757130 | 60757130 | C/A | 1.8 × 10−11 | 2.9 ± 1.17 | 1.8 × 10−11 | 3.0 ± 1.8 | 6.4 × 10−11 | 0.026 | 2.63 × 10−10 | 0.49 | 0.95 | 1.0 | 0.8 |
| 2 | 2:60750303 | 60750303 | T/C | 4.4 × 10−11 | 2.8 ± 1.16 | 4.4 × 10−11 | 2.8 ± 1.2 | 1.7 × 10−10 | 0.009 | 1.6 × 10−10 | 0.32 | 0.96 | 1.0 | 0.8 |
| 2 | 2:60697654 | 60697654 | A/C | 6.7 × 10−11 | 2.4 ± 1.14 | 6.7 × 10−11 | 2.4 ± 1.1 | 1.5 × 10−10 | 0.039 | 3.4 × 10−10 | 0.88 | 0.93 | 1.0 | 0.7 |
| 2 | 2:60755762 | 60755762 | T/C | 7.1 × 10−11 | 2.8 ± 1.17 | 7.1 × 10−11 | 2.8 ± 1.2 | 2.0 × 10−10 | 0.011 | 1.2 × 10−9 | 0.84 | 0.97 | 1.0 | 0.7 |
| 2 | 2:60756755 | 60756755 | G/C | 7.4 × 10−11 | 2.8 ± 1.17 | 7.4 × 10−11 | 2.8 ± 1.2 | 2.3 × 10−10 | 0.016 | 1.2 × 10−9 | 0.65 | 0.9 | 1.0 | 0.8 |
| 2 | rs575474598 | 60710738 | G/A | 9.0 × 10−11 | 0.4 ± 1.14 | 0.32 | 0.5 ± 2.2 | 1.7 × 10−16 | 9.03 × 10−5 | 2.4 × 10−14 | 0.01 | 0.9 | 1.0 | 0.0 |
| 2 | rs1236323224 | 60756504 | T/C | 1.3 × 10−10 | 2.8 ± 1.17 | 1.3 × 10−10 | 2.8 ± 1.2 | 4.3 × 10−10 | 0.016 | 2.05 × 10−9 | 0.72 | 0.9 | 1.0 | 0.8 |
| 2 | 2:60736852 | 60736852 | A/G | 1.8 × 10−10 | 2.6 ± 1.15 | 7.7 × 10−5 | 2.6 ± 1.3 | 4.2 × 10−10 | 0.002 | 6.4 × 10−9 | 0.41 | 0.9 | 1.0 | 0.7 |
| 2 | 2:60743605 | 60743605 | G/A | 3.6 × 10−10 | 2.5 ± 1.15 | 0.005 | 2.4 ± 1.4 | 2.2 × 10−10 | 0.005 | 1.5 × 10−9 | 0.51 | 0.9 | 1.0 | 0.5 |
| 2 | 2:60729702 | 60729702 | G/A | 1.1 × 10−8 | 2.4 ± 1.16 | 0.26 | 1.7 ± 1.7 | 4.6 × 10−9 | 0.011 | 1.9 × 10−8 | 0.15 | 0.9 | 1.0 | 0.3 |
| 2 | 2:60684034 | 60684034 | C/T | 4.8 × 10−7 | 3.1 ± 1.25 | 4.8 × 10−7 | 3.1 ± 1.3 | 2.0 × 10−6 | 0.054 | 6.1 × 10−6 | 0.17 | 0.9 | 1.0 | 0.8 |
| 2 | 2:60698461 | 60698461 | T/C | 5.9 × 10−7 | 1.9 ± 1.13 | 0.1 | 1.6 ± 1.4 | 1.2 × 10−7 | 0.092 | 1.06 × 10−7 | 0.7 | 0.8 | 1.0 | 0.2 |
| 2 | 2:60708597 | 60708597 | C/T | 9.2 × 10−7 | 0.5 ± 1.13 | 0.4 | 0.6 ± 1.8 | 1.1 × 10−9 | 0.004 | 8.01 × 10−9 | 0.02 | 0.9 | 1.0 | 0.004 |
| 2 | 2:60701335 | 60701335 | C/T | 1.4 × 10−6 | 1.8 ± 1.13 | 0.3 | 1.4 ± 1.4 | 1.2 × 10−7 | 0.6 | 2.8 × 10−8 | 0.57 | 0.4 | 1.0 | 0.1 |
| 2 | 2:60698397 | 60698397 | C/A | 3.8 × 10−6 | 1.8 ± 1.13 | 0.6 | 1.3 ± 1.8 | 3.7 × 10−8 | 0.06 | 4.3 × 10−8 | 0.01 | 0.8 | 1.0 | 0.01 |
| 2 | 2:60679942 | 60679942 | C/T | 4.5 × 10−6 | 0.5 ± 1.18 | 3.7 × 10−5 | 0.5 ± 1.2 | 2.1 × 10−5 | 0.01 | 0.0002 | 0.04 | 0.9 | 0.9 | 0.9 |
| 2 | 2:60748758 | 60748758 | G/A | 5.07 × 10−6 | 0.5 ± 1.14 | 5.0 × 10−6 | 0.6 ± 1.1 | 9.1 × 10−6 | 0.43 | 3.5 × 10−6 | 0.9 | 0.6 | 1.0 | 0.6 |
| 2 | 2:60696528 | 60696528 | C/T | 1.6 × 10−5 | 2.2 ± 1.2 | 0.06 | 2.4 ± 1.6 | 1.2 × 10−6 | 0.02 | 2.2 × 10−6 | 0.82 | 0.8 | 1.0 | 0.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ginete, C.; Delgadinho, M.; Santos, B.; Miranda, A.; Silva, C.; Guerreiro, P.; Chimusa, E.R.; Brito, M. Genetic Modifiers of Sickle Cell Anemia Phenotype in a Cohort of Angolan Children. Genes 2024, 15, 469. https://doi.org/10.3390/genes15040469
Ginete C, Delgadinho M, Santos B, Miranda A, Silva C, Guerreiro P, Chimusa ER, Brito M. Genetic Modifiers of Sickle Cell Anemia Phenotype in a Cohort of Angolan Children. Genes. 2024; 15(4):469. https://doi.org/10.3390/genes15040469
Chicago/Turabian StyleGinete, Catarina, Mariana Delgadinho, Brígida Santos, Armandina Miranda, Carina Silva, Paulo Guerreiro, Emile R. Chimusa, and Miguel Brito. 2024. "Genetic Modifiers of Sickle Cell Anemia Phenotype in a Cohort of Angolan Children" Genes 15, no. 4: 469. https://doi.org/10.3390/genes15040469
APA StyleGinete, C., Delgadinho, M., Santos, B., Miranda, A., Silva, C., Guerreiro, P., Chimusa, E. R., & Brito, M. (2024). Genetic Modifiers of Sickle Cell Anemia Phenotype in a Cohort of Angolan Children. Genes, 15(4), 469. https://doi.org/10.3390/genes15040469