

Recessive GNE Mutations in Korean Nonaka Distal Myopathy Patients with or without Peripheral Neuropathy

Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Clinical and Electrophysiological Examinations
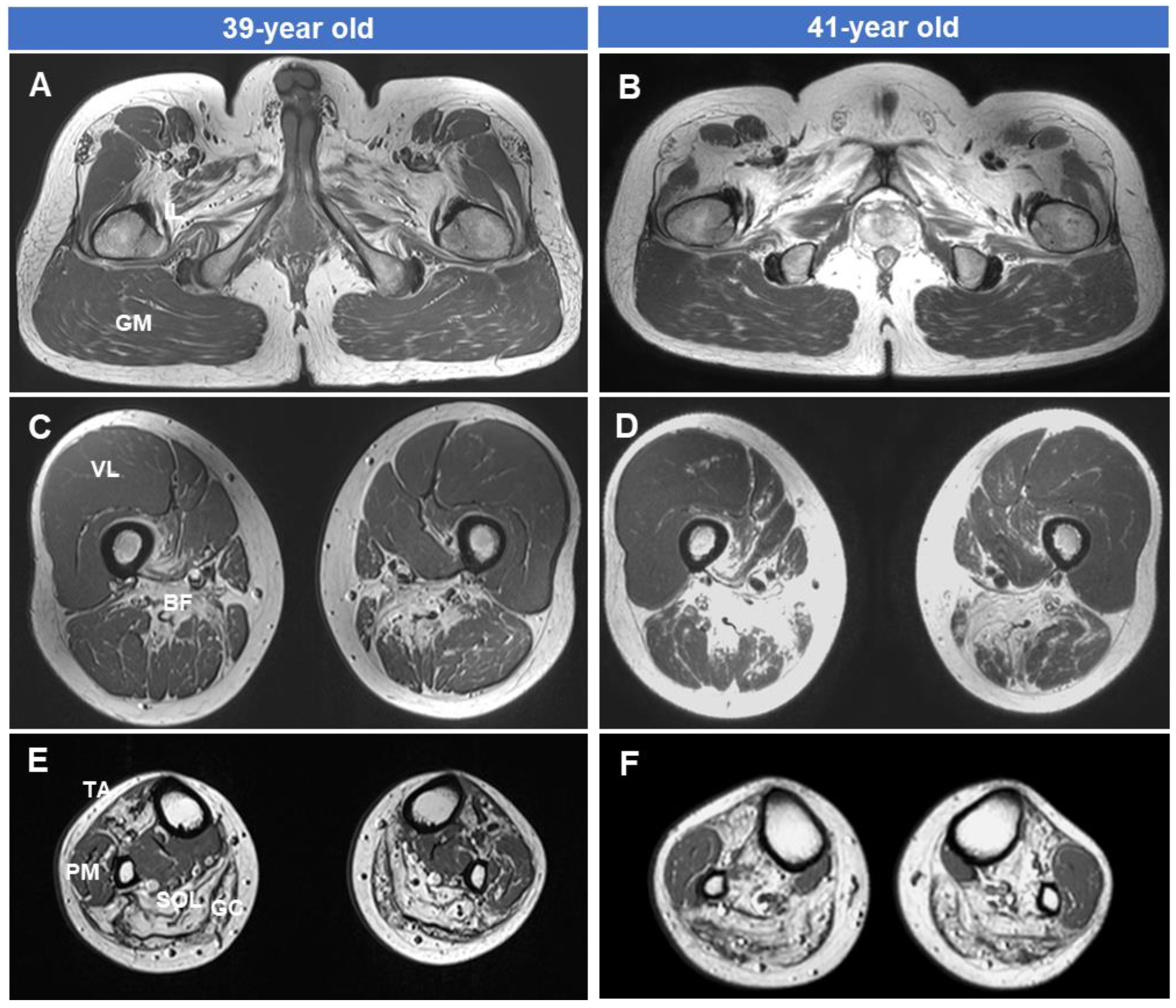
2.3. Lower Extremity MRI
2.4. Nucleic Acid Extraction and Exome Sequencing
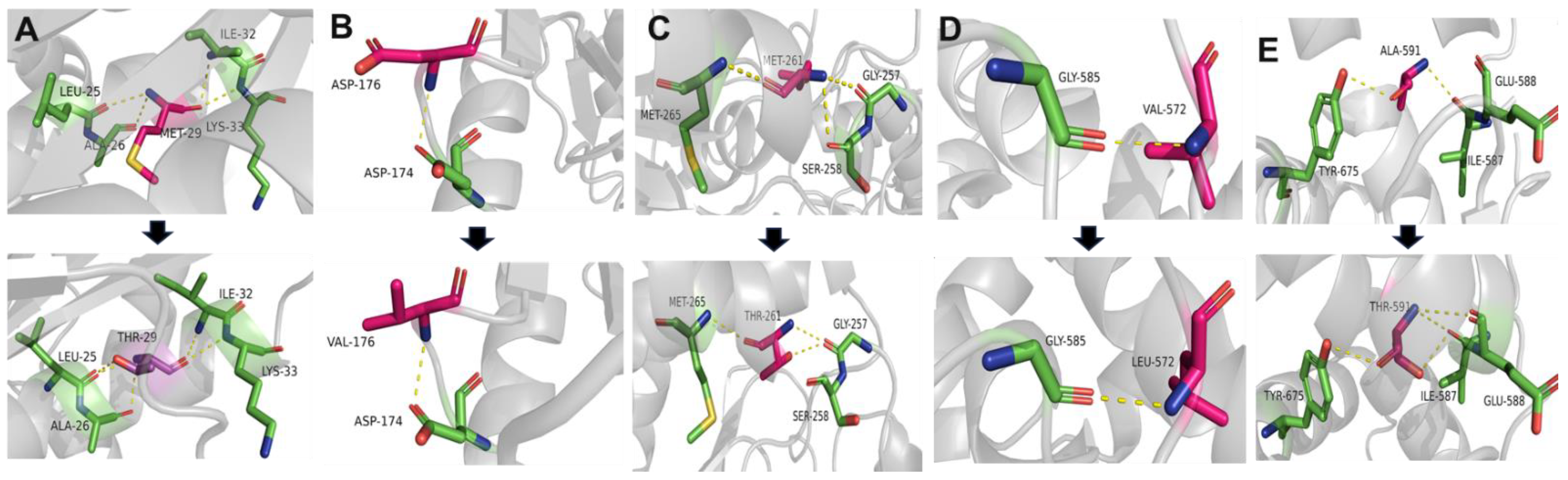
2.5. Conservation and Prediction of 3D Structural Changes of GNE Variants
3. Results
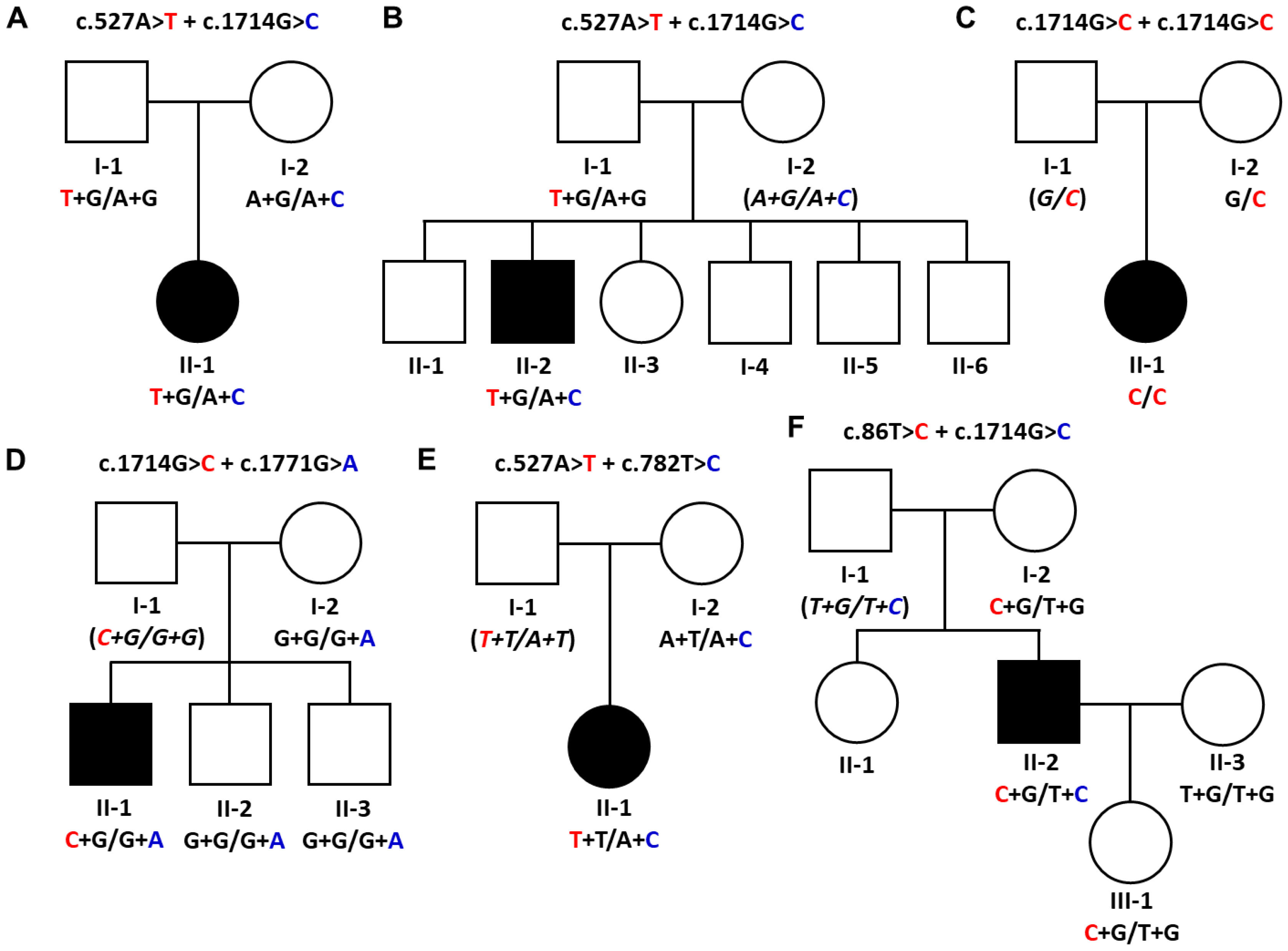
3.1. GNE Mutations as the Underlying Causes of Myopathy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family ID | Nucleotide Change 1 | Amino Acid Change 1 | Main Phenotype | Mutant Allele Frequency | ACMG/AMP | References | |||
|---|---|---|---|---|---|---|---|---|---|
| IGSR | gnomAD | ESP | KRGDB | ||||||
| Myo-6 | c.527A>T c.1714G>C | p.Asp176Val p.Val572Leu | DMRV | 0.0004 0.0002 | 0.000495 0.000016 | URUR | 0.002038 0.002037 | P P | [30,31] |
| Myo-8 | c.527A>T c.1714G>C | p.Asp176Val p.Val572Leu | Distal myopathy | 0.0004 0.0002 | 0.000495 0.000016 | URUR | 0.002038 0.002037 | P P | [30,31] |
| Myo-12 | c.1714G>C c.1714G>C | p.Val572Leu p.Val572Leu | Distal myopathy | 0.0002 | 0.000016 | UR | 0.002037 | P | [16,34] |
| Myo-24 | c.1714G>C c.1771G>A | p.Val572Leu p.Ala591Thr | Distal myopathy | 0.0002 UR | 0.000016 UR | UR UR | 0.002037 UR | P LP | [15,35] |
| Myo-35 | c.527A>T c.782T>C | p.Asp176Val p.Met261Thr | Distal myopathy | 0.0004 UR | 0.000495 UR | UR UR | 0.002038 UR | P LP | This study 2 |
| Myo-41 | c.86T>C c.1714G>C | p.Met29Thr p.Val572Leu | Distal myopathy and dHMN | UR 0.0002 | UR 0.000016 | UR UR | UR 0.002037 | P LP | This study 3 |
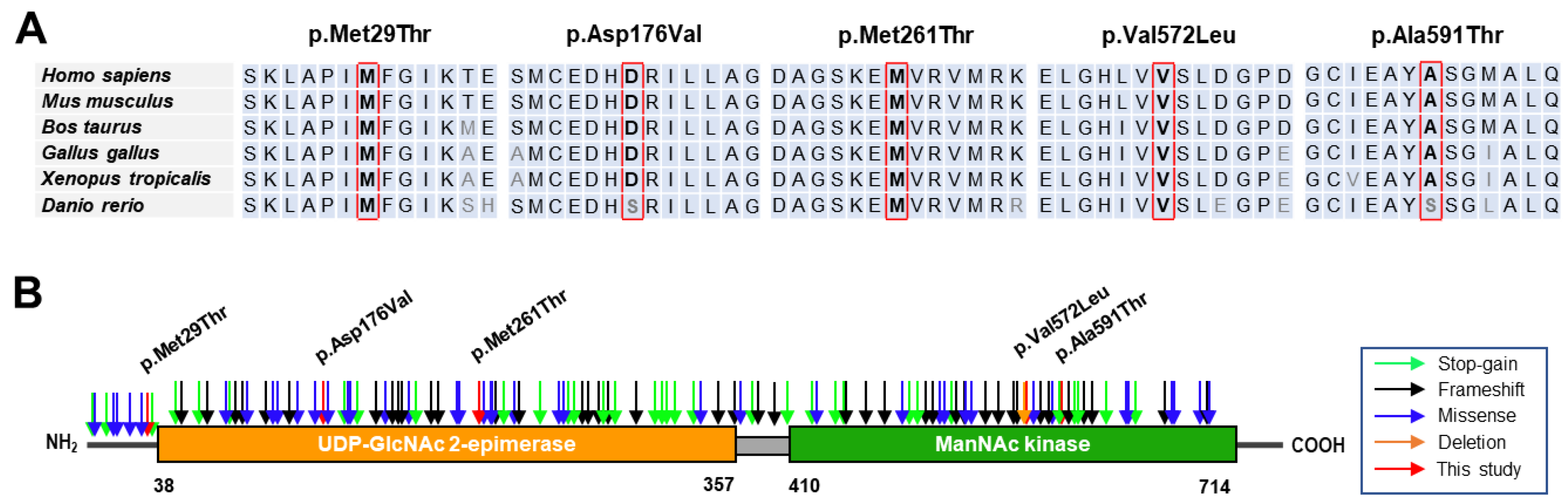
3.2. Conservation Analysis and In Silico Prediction of the Mutation Effects
3.3. Clinical Manifestations
3.4. Fatty Replacements of Lower Extremity Muscles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carrillo, N.; Malicdan, M.C.; Huizing, M. GNE myopathy: Etiology, diagnosis, and therapeutic challenges. Neurotherapeutics 2018, 15, 900–914. [Google Scholar] [CrossRef]
- Mullen, J.; Alrasheed, K.; Mozaffar, T. GNE myopathy: History, etiology, and treatment trials. Front. Neurol. 2022, 13, 1002310. [Google Scholar] [CrossRef]
- Nishino, I.; Carrillo-Carrasco, N.; Argov, Z. GNE myopathy: Current update and future therapy. J. Neurol. Neurosurg. Psychiatry 2015, 86, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.; Carrillo-Carrasco, N.; Malicdan, M.C.; Noguchi, S.; Gahl, W.A.; Mitrani-Rosenbanum, S.; Argov, Z.; Nishino, I. GNE myopathy: New name and new mutation nomenclature. Neuromuscul. Disord. 2014, 24, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Mori-Yoshimura, M.; Oya, Y.; Yajima, H.; Yonemoto, N.; Kobayashi, Y.; Hayashi, Y.K.; Noguchi, S.; Nishino, I.; Murata, M. GNE myopathy: A prospective natural history study of disease progression. Neuromuscul. Disord. 2014, 24, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Pogoryelova, O.; González Coraspe, J.A.; Nikolenko, N.; Lochmüller, H.; Roos, A. GNE myopathy: From clinics and genetics to pathology and research strategies. Orphanet J. Rare Dis. 2018, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, I.; Sunohara, N.; Ishiura, S.; Satoyoshi, E. Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J. Neurol. Sci. 1981, 51, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Mitrani-Rosenbaum, S.; Argov, Z.; Blumenfeld, A.; Sediman, C.E.; Sediman, J.G. Hereditary inclusion body myopathy maps to chromosome 9p1-q1. Hum. Mol. Genet. 1996, 5, 159–163. [Google Scholar] [CrossRef]
- Asaka, T.; Ikeuchi, K.; Okino, S.; Takizawa, Y.; Satake, R.; Nitta, E.; Komai, K.; Endo, K.; Higuchi, S.; Oyake, T.; et al. Homozygosity and linkage disequilibrium mapping of autosomal recessive distal myopathy (Nonaka distal myopathy). J. Hum. Genet. 2001, 46, 649–655. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eisenberg, I.; Avidan, N.; Potikha, T.; Hochner, H.; Chen, M.; Olender, T.; Barash, M.; Shemesh, M.; Sadeh, M.; Grabov-Nardini, G.; et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat. Genet. 2001, 29, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Kayashima, T.; Matsuo, H.; Satoh, A.; Ohta, T.; Yoshiura, K.; Matsumoto, N.; Nakane, Y.; Niikawa, N.; Kishino, T. Nonaka myopathy is caused by mutations in the UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase gene (GNE). J. Hum. Genet. 2002, 47, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Broccolini, A.; Pescatori, M.; D’Amico, A.; Sabino, A.; Silvestri, G.; Ricci, E.; Servidei, S.; Tonali, P.A.; Mirabella, M. An Italian family with autosomal recessive inclusion-body myopathy and mutations in the GNE gene. Neurology 2002, 59, 1808–1809. [Google Scholar] [CrossRef]
- Cerino, M.; Gorokhova, S.; Béhin, A.; Urtizberea, J.A.; Kergourlay, V.; Salvo, E.; Bernard, R.; Levy, N.; Bartoli, M.; Krahn, M. Novel pathogenic variants in a French cohort widen the mutational spectrum of GNE myopathy. J. Neuromuscul. Dis. 2015, 2, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Pogoryelova, O.; Cammish, P.; Mansbach, H.; Argov, Z.; Nishino, I.; Skrinar, A.; Chan, Y.; Nafissi, H.; Kakkis, E.; Lochmuller, H. Phenotypic stratification and genotype-phenotype correlation in a heterogeneous, international cohort of GNE myopathy patients: First report from the GNE myopathy Disease Monitoring Program, registry portion. Neuromuscul. Disord. 2018, 28, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Ki, C.S.; Kim, J.W.; Sung, D.H.; Choi, Y.C.; Kim, S.H. Mutation analysis of the GNE gene in Korean patients with distal myopathy with rimmed vacuoles. J. Hum. Genet. 2006, 51, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.E.; Park, H.-J.; Shin, H.Y.; Nam, T.-S.; Kim, S.M.; Choi, Y.-C. Clinical characteristics and molecular genetic analysis of Korean patients with GNE myopathy. Yonsei Med. J. 2013, 54, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.E.; Kim, D.-S.; Choi, Y.C.; Shin, J.H. Progression of GNE myopathy based on the patient-reported outcome. J. Clin. Neurol. 2019, 15, 275–284. [Google Scholar] [CrossRef]
- Tanner, M.E. The enzymes of sialic acid biosynthesis. Bioorg. Chem. 2005, 33, 216–228. [Google Scholar] [CrossRef]
- Hinderlich, S.; Weidemann, W.; Yardeni, T.; Horstkorte, R.; Huizing, M. UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE): A master regulator of sialic acid synthesis. Top. Curr. Chem. 2015, 366, 97–137. [Google Scholar]
- Awasthi, K.; Srivastava, A.; Bhattacharya, S.; Bhattacharya, A. Tissue specific expression of sialic acid metabolic pathway: Role in GNE myopathy. J. Muscle Res. Cell Motil. 2001, 42, 99–116. [Google Scholar] [CrossRef]
- Malicdan, M.C.V.; Noguchi, S.; Nonaka, I.; Hayashi, Y.K.; Nishino, I. A Gne knockout mouse expressing human V572L mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum. Mol. Genet. 2007, 16, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Malicdan, M.C.V.; Noguchi, S.; Nonaka, I.; Hayashi, Y.K.; Nishino, I. A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum. Mol. Genet. 2007, 16, 2669–2682. [Google Scholar] [CrossRef] [PubMed]
- Previtali, S.C.; Zhao, E.; Lazarevic, D.; Pipitone, G.B.; Fabrizi, G.M.; Manganelli, F.; Mazzeo, A.; Pareyson, D.; Schenone, A.; Taroni, F.; et al. Expanding the spectrum of genes responsible for hereditary motor neuropathies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1171–1179. [Google Scholar] [CrossRef]
- Grecu, N.; Villa, L.; Cavalli, M.; Ristaino, A.; Choumert, A.; Butori, C.; Salviati, L.; Puma, A.; Krahn, M.; Cerino, M.; et al. Motor axonal neuropathy associated with GNE mutations. Muscle Nerve 2021, 63, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.Y.; Duan, H.Q.; Li, Q.X.; Luo, Y.B.; Bi, F.F.; Huang, K.; Yang, H. Expanding the clinicopathological-genetic spectrum of GNE myopathy by a Chinese neuromuscular centre. J. Cell Mol. Med. 2021, 25, 10494–10503. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, H.; Lin, Z.; Li, X.; Liu, L.; Huang, S.; Zhao, H.; Zhu, X.; Xiao, Q.; Duan, R.; et al. The genetic and clinical spectrum in a cohort of 39 families with complex inherited peripheral neuropathies. J. Neurol. 2023, 270, 4959–4967. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Kanwal, S.; Hameed, R.; Tamanna, N.; Perveen, S.; Mahreen, H.; Son, W.; Lee, K.S.; Chung, K.W. Biallelic mutations in pakistani families with autosomal recessive prelingual nonsyndromic hearing loss. Genes Genom. 2023, 45, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Rosignoli, S.; Paiardini, A. Boosting the full potential of PyMOL with structural biology plugins. Biomolecules 2022, 12, 1764. [Google Scholar] [CrossRef]
- Seo, S.D.; Park, H.J.; Song, H.S.; Kim, H.J.; Park, J.M.; Hong, Y.B.; Chung, K.W.; Choi, B.O. Distal myopathy with rimmed vacuoles confirmed by whole exome sequencing. J. Life Sci. 2014, 24, 311–317. [Google Scholar] [CrossRef]
- Choi, Y.A.; Park, S.H.; Yi, Y.; Kim, K. Novel mutation of the GNE gene presenting atypical mild clinical feature: A Korean case report. Ann. Rehabil. Med. 2015, 39, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Arai, A.; Tanaka, K.; Ikeuchi, T.; Igarashi, S.; Kobayashi, H.; Asaka, T.; Date, H.; Saito, M.; Tanaka, H.; Kawasaki, S.; et al. A novel mutation in the GNE gene and a linkage disequilibrium in Japanese pedigrees. Ann. Neurol. 2002, 52, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Toriumi, Y.; Takusa, Y.; Uchiyama, A.; Kimura, M.; Sejima, H.; Yamaguchi, S.; Eda, I.; Nishino, I.; Nonaka, I. Distal myopathy with rimmed vacuoles in a case of opercular syndrome. Brain Dev. 2006, 28, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.E.; Kim, H.S.; Choi, E.S.; Shin, J.H.; Kim, S.Y.; Son, E.H.; Lee, C.H.; Kim, D.S. Limb-girdle phenotype is frequent in patients with myopathy associated with GNE mutations. J. Neurol. Sci. 2012, 321, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Shin, J.H.; Park, J.S. GNE myopathy with prominent axial muscle involvement. J. Clin. Neurol. 2018, 14, 580–582. [Google Scholar] [CrossRef]
- Li, H.; Chen, Q.; Liu, F.; Zhang, X.; Liu, T.; Li, W.; Liu, S.; Zhao, Y.; Wen, B.; Dai, T.; et al. Clinical and molecular genetic analysis in Chinese patients with distal myopathy with rimmed vacuoles. J. Hum. Genet. 2011, 56, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Pu, C.; Huang, X.; Liu, J.; Mao, Y. Distal myopathy with rimmed vacuoles: Clinical and muscle morphological characteristics and spectrum of GNE gene mutations in 53 Chinese patients. Neurol. Res. 2011, 33, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Mori-Yoshimura, M.; Monma, K.; Suzuki, N.; Aoki, M.; Kumamoto, T.; Tanaka, K.; Tomimitsu, H.; Nakano, S.; Sonoo, M.; Shimizu, J.; et al. Heterozygous UDP-GlcNAc 2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE gene result in a less severe GNE myopathy phenotype compared to homozygous N-acetylmannosamine kinase domain mutations. Neurol. Sci. 2012, 318, 100–105. [Google Scholar] [CrossRef]
- Cho, A.; Hayashi, Y.K.; Monma, K.; Oya, Y.; Noguchi, S.; Nonaka, I.; Nishino, I. Mutation profile of the GNE gene in Japanese patients with distal myopathy with rimmed vacuoles (GNE myopathy). J. Neurol. Neurosurg. Psychiatry 2014, 85, 914–917. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Z.; Hong, D.; Lv, H.; Zhang, W.; Chen, J.; Yuan, Y. Mutational spectrum and clinical features in 35 unrelated mainland Chinese patients with GNE myopathy. J. Neurol. Sci. 2015, 354, 21–26. [Google Scholar] [CrossRef]
- Argov, Z.; Eisenberg, I.; Grabov-Nardini, G.; Sadeh, M.; Wirguin, I.; Soffer, D.; Mitrani-Rosenbaum, S. Hereditary inclusion body myopathy: The Middle Eastern genetic cluster. Neurology 2003, 60, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Khadilkar, S.V.; Nalini, A.; Ganapathy, A.; Mannan, A.U.; Majumder, P.P.; Bhattacharya, A. Mutation spectrum of GNE myopathy in the Indian sub-continent. J. Neuromuscul. Dis. 2018, 5, 85–92. [Google Scholar] [CrossRef]
- Murtazina, A.; Nikitin, S.; Rudenskaya, G.; Sharkova, I.; Borovikov, A.; Sparber, P.; Shchagina, O.; Chukhrova, A.; Ryzhkova, O.; Shatokhina, O.; et al. Genetic and clinical spectrum of GNE myopathy in Russia. Genes 2022, 13, 1991. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Hong, Y.B.; Choi, Y.C.; Lee, J.; Kim, E.J.; Lee, J.S.; Mo, W.M.; Ki, H.I.; Kim, H.J.; Hyun, Y.S.; et al. ADSSL1 mutation relevant to autosomal recessive adolescent onset distal myopathy. Ann. Neurol. 2016, 79, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Nam, S.H.; Park, J.M.; Kanwal, S.; Choi, Y.J.; Lee, H.J.; Lee, J.E.; Park, J.S.; Choi, B.O.; Chung, K.W. Compound heterozygous mutations of SH3TC2 in Charcot-Marie-Tooth disease type 4C patients. J. Hum. Genet. 2019, 64, 961–965. [Google Scholar] [CrossRef]
- Mori-Yoshimura, M.; Oya, Y.; Hayashi, Y.K.; Noguchi, S.; Nishino, I.; Murata, M. Respiratory dysfunction in patients severely affected by GNE myopathy (distal myopathy with rimmed vacuoles). Neuromuscul. Disord. 2013, 23, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.K.; Kim, E.J.; Ki, C.S.; Kim, J.W. Nonaka Myopathy: A case report. J. Korean Acad. Rehabil. Med. 2004, 28, 288–291. [Google Scholar]
- Han, Y.S.; Kim, D.E.; Kim, J.M.; Cho, J.S.; Han, J.H.; Cho, E.K.; Ki, C.S.; Kim, J.W. A case of Nonaka myopathy confirmed by GNE mutation. J. Korean Neurol. Assoc. 2005, 23, 418–421. [Google Scholar]
- Chaouch, A.; Brennan, K.M.; Hudson, J.; Longman, C.; McConville, J.; Morrison, P.J.; Farrugia, M.E.; Petty, R.; Stewart, W.; Norwood, F.; et al. Two recurrent mutations are associated with GNE myopathy in the North of Britain. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1359–1365. [Google Scholar] [CrossRef]
- Yoshioka, W.; Nishino, I.; Noguchi, S. Recent advances in establishing a cure for GNE myopathy. Curr. Opin. Neurol. 2022, 35, 629–636. [Google Scholar] [CrossRef]
- Carrillo, N.; Malicdan, M.C.; Leoyklang, P.; Shrader, J.A.; Joe, G.; Slota, C.; Perreault, J.; Heiss, J.D.; Class, B.; Liu, C.U.; et al. Safety and efficacy of N-acetylmannosamine (ManNAc) in patients with GNE myopathy: An open-label phase 2 study. Genet. Med. 2021, 23, 2067–2075. [Google Scholar] [CrossRef]
- Park, Y.E.; Park, E.; Choi, J.; Go, H.; Park, D.B.; Kim, M.-Y.; Sung, N.J.; Kim, L.; Shin, J.H. Pharmacokinetics and clinical efficacy of 6′-sialyllactose in patients with GNE myopathy: Randomized pilot trial. Biomed. Pharmacother. 2023, 168, 115689. [Google Scholar] [CrossRef] [PubMed]
- Park, J.C.; Kim, J.; Jang, H.K.; Lee, S.-Y.; Kim, K.T.; Kwon, E.J.; Park, S.; Lee, H.S.; Choi, H.; Park, S.Y.; et al. Multiple isogenic GNE-myopathy modeling with mutation specific phenotypes from human pluripotent stem cells by base editors. Biomaterials 2022, 282, 121419. [Google Scholar] [CrossRef] [PubMed]




| Variant 1 | Domain | dbSNP | GERP | In silico Prediction 2 | ||||
|---|---|---|---|---|---|---|---|---|
| Nucleotide | Amino Acid | PP2 | MutT | MUp | REVEL | |||
| c.86T>C | p.Met29Thr | Epimerase | UR | 4.83 | 0.983 * | 1.00 * | −1.00 * | 0.93 * |
| c.527A>T | p.Asp176Val | Epimerase | rs139425890 | 5.67 | 0.108 | 0.99 * | −0.11 * | 0.80 * |
| c.782T>C | p.Met261Thr | Epimerase | UR | 5.77 | 0.996 * | 0.94 * | −1.00 * | 0.84 * |
| c.1714G>C | p.Val572Leu | Kinase | rs121908632 | 5.75 | 0.968 * | 0.96 * | −0.36 * | 0.83 * |
| c.1771G>A | p.Ala591Thr | Kinase | rs752286512 | 5.75 | 0.957 * | 0.94 * | −0.38 * | 0.77 * |
| Family: Patient | Myo-6: II-1 | Myo-8: II-2 | Myo-12: II-1 | Myo-24: II-1 | Myo-35: II-1 | Myo-41: III-1 |
|---|---|---|---|---|---|---|
| Sex | Female | Male | Female | Male | Female | Male |
| Mutation | p.Asp176Val p.Val572Leu | p.Asp176Val p.Val572Leu | p.Val572Leu p.Val572Leu | p.Val572Leu p.Ala591Thr | p.Asp176Val p.Met261Thr | p.Met29Thr p.Val572Leu |
| Examined age (yrs) | 38 | 48 | 25 | 23 | 29 | 38 |
| Onset–leg (yrs) | 35 | 38 | 20 | 19 | 25 | 34 |
| Onset–hand (yrs) | 36 | 40 | 22 | 19 | - | 37 |
| Family history | No | No | No | No | No | No |
| Affected muscle | LE/DM → UE | LE/DM → PM/UE | LE/DM&PM → UE | LE&UE/DM | LE/DM&PM | LE/DM → UE |
| Electromyography | Myopathy | Myopathy | Myopathy | Myopathy | Myopathy | Myopathy, neuropathy |
| Phenotype | Distal myopathy | Distal myopathy | Distal myopathy | Distal myopathy | Distal myopathy | Distal myopathy, dHMN |
| Sensory loss | No | No | No | No | No | No |
| Knee jerk (Right/Left) | +/+ | −/− | +/+ | +/+ | −/− | −/− |
| Foot drop | Yes | Yes | Yes | Yes | Yes | Yes |
| Core muscles involved | No | Yes 1 | No | No | No | No |
| Wheelchair-bound | No | Yes | No | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamanna, N.; Pi, B.K.; Lee, A.J.; Kanwal, S.; Choi, B.-O.; Chung, K.W. Recessive GNE Mutations in Korean Nonaka Distal Myopathy Patients with or without Peripheral Neuropathy. Genes 2024, 15, 485. https://doi.org/10.3390/genes15040485
Tamanna N, Pi BK, Lee AJ, Kanwal S, Choi B-O, Chung KW. Recessive GNE Mutations in Korean Nonaka Distal Myopathy Patients with or without Peripheral Neuropathy. Genes. 2024; 15(4):485. https://doi.org/10.3390/genes15040485
Chicago/Turabian StyleTamanna, Nasrin, Byung Kwon Pi, Ah Jin Lee, Sumaira Kanwal, Byung-Ok Choi, and Ki Wha Chung. 2024. "Recessive GNE Mutations in Korean Nonaka Distal Myopathy Patients with or without Peripheral Neuropathy" Genes 15, no. 4: 485. https://doi.org/10.3390/genes15040485
APA StyleTamanna, N., Pi, B. K., Lee, A. J., Kanwal, S., Choi, B.-O., & Chung, K. W. (2024). Recessive GNE Mutations in Korean Nonaka Distal Myopathy Patients with or without Peripheral Neuropathy. Genes, 15(4), 485. https://doi.org/10.3390/genes15040485