
Population Genomics of Commercial Fish Sebastes schlegelii of the Bohai and Yellow Seas (China) Using a Large SNP Panel from GBS



Abstract
:1. Introduction
2. Materials and Methods
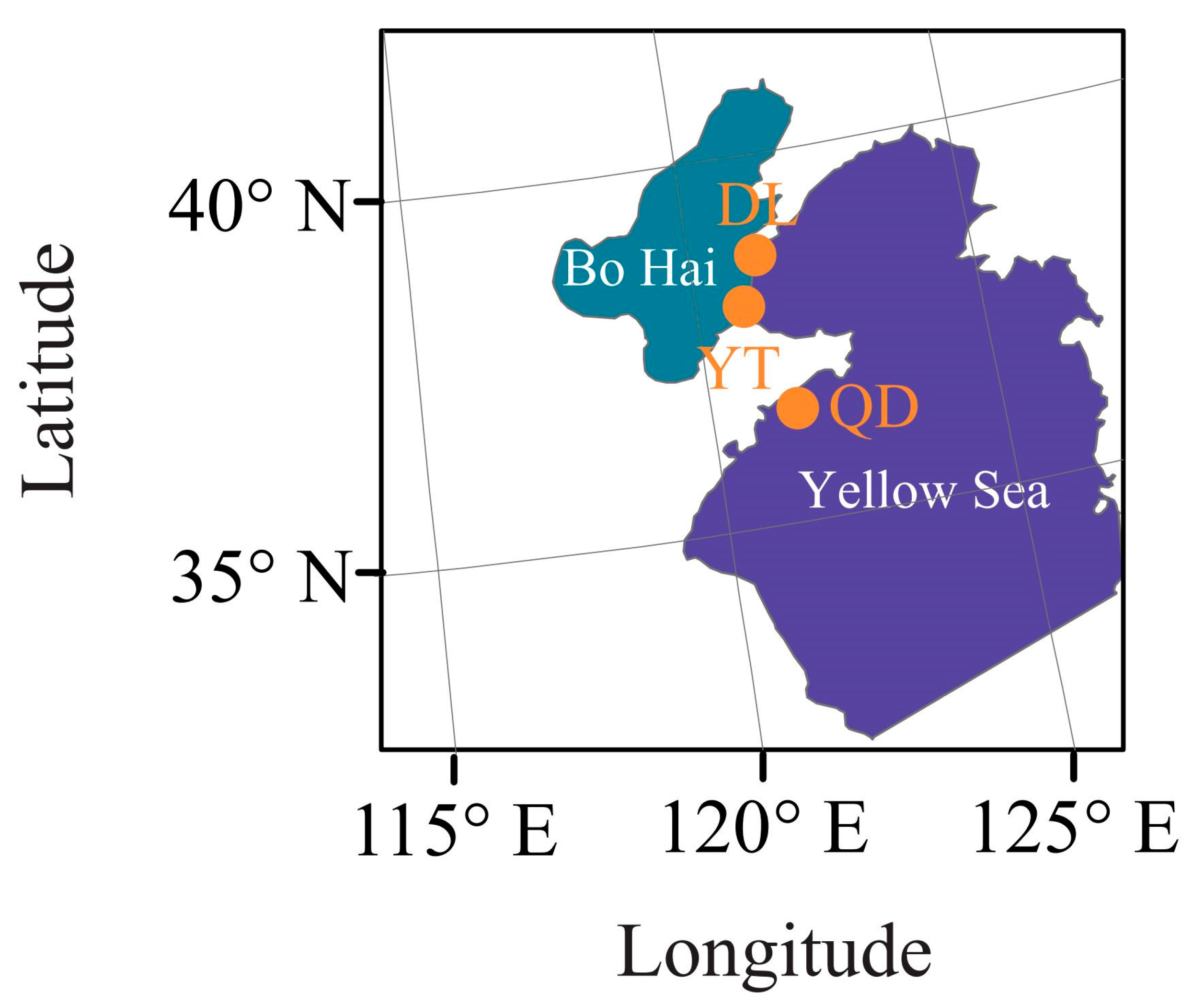
2.1. Sample Collection and DNA Extraction
2.2. GBS Library Construction, Illumina Sequencing, and Read Mapping
2.3. Variant Calling, Population Structure, Diversity, and Divergence
2.4. Selection Pressure Analysis
3. Results
3.1. Sequencing and Variants Calling
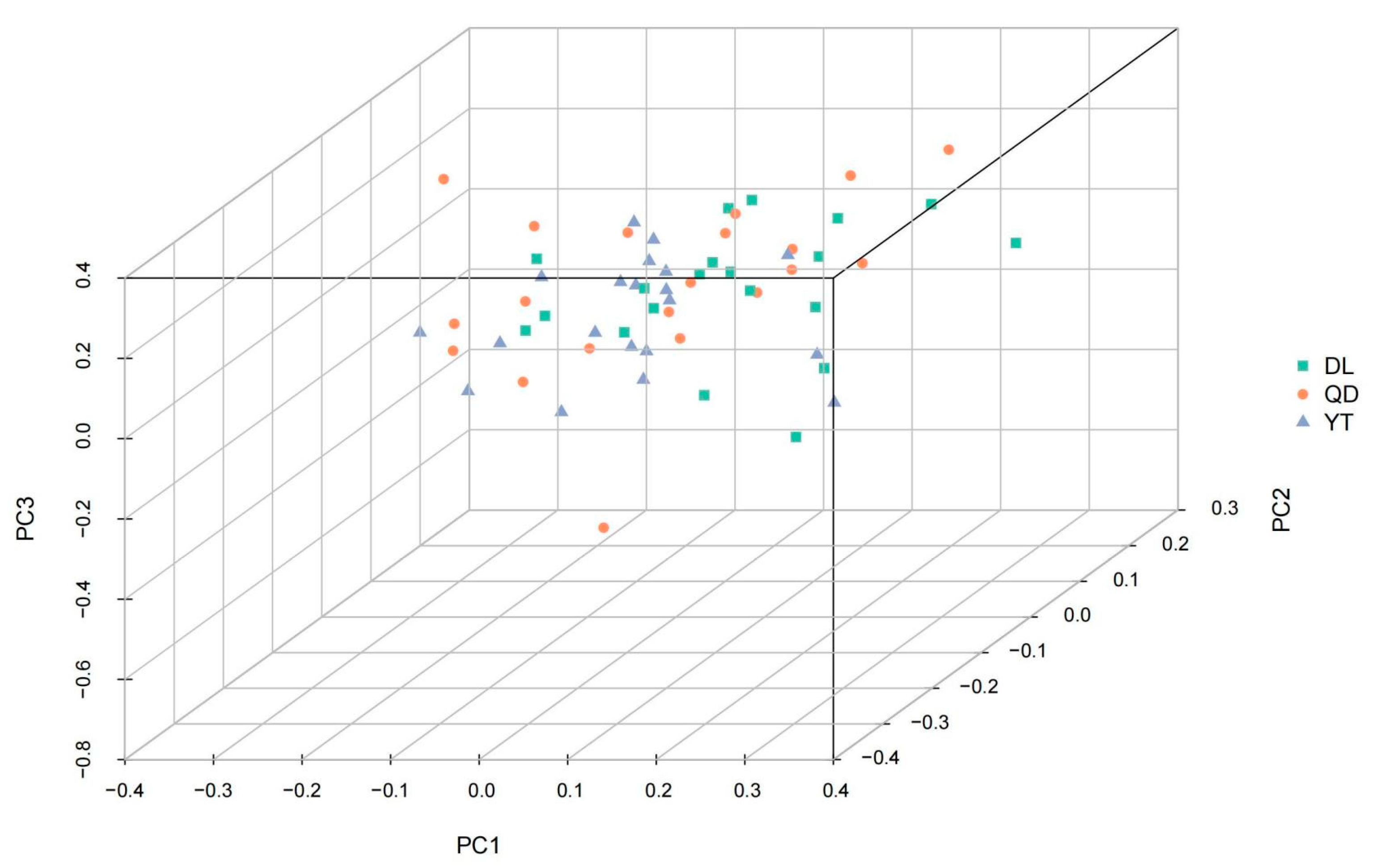
3.2. Population Structure of S. schlegelii
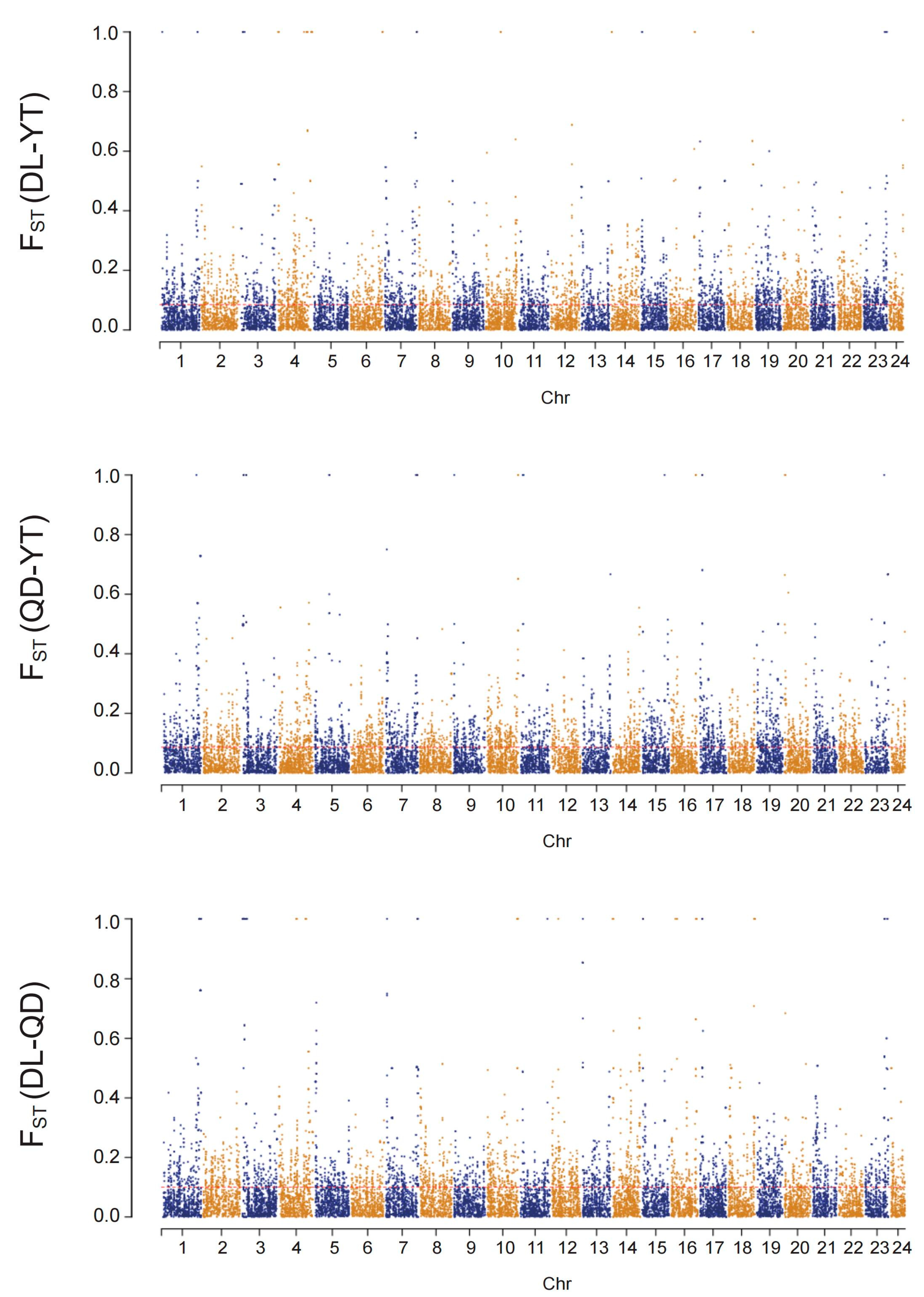
3.3. Genetic Diversity and Divergence of S. schlegelii Population
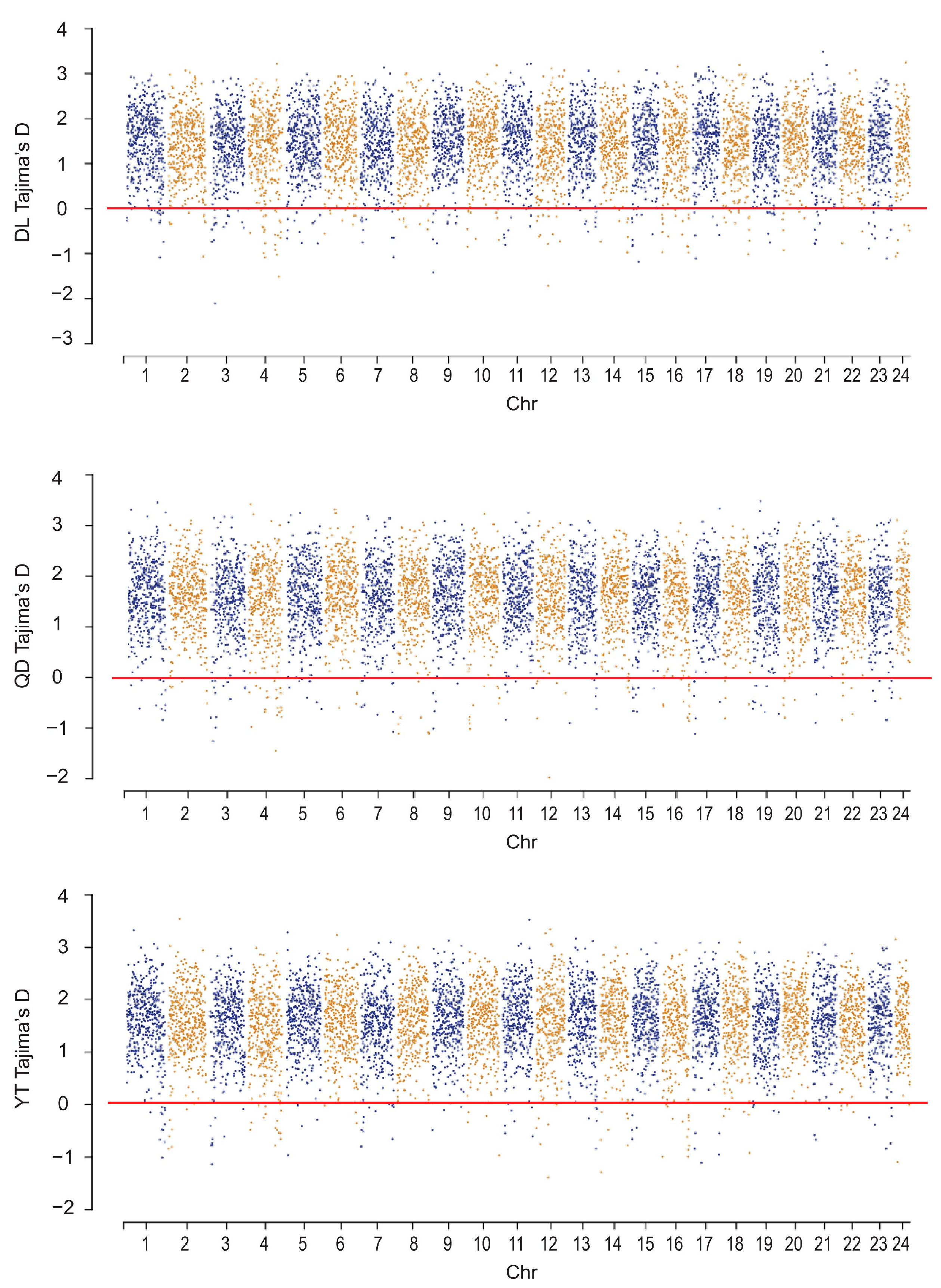
3.4. Genome-Wide Selection Pressure Analysis
4. Discussion
4.1. Genetic Differentiation Analysis
4.2. Genetic Diversity and Selection Pressure Analysis
4.3. Population Structure and Environmental Adaptation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Song, W.; Xie, Y.; Sun, M.; Li, X.; Fitzpatrick, C.K.; Vaux, F.; O’Malley, K.G.; Zhang, Q.; Qi, J.; He, Y. A duplicated amh is the master sex-determining gene for Sebastes rockfish in the Northwest Pacific. Open Biol. 2021, 11, 210063. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, H.; Zhang, Q.; Zhang, H.; Zhao, J. Trophic interactions of reef-associated predatory fishes (Hexagrammos otakii and Sebastes schlegelii) in natural and artificial reefs along the coast of North Yellow Sea. China Sci. Total Environ. 2021, 791, 148250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cao, M.; Xiu, Y.; Fu, Q.; Yang, N.; Su, B.; Li, C. Identification of antimicrobial peptide genes in black rockfish Sebastes schlegelii and their responsive mechanisms to Edwardsiella tarda infection. Biology 2021, 10, 1015. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Chen, B.; Xia, B.; Shi, X.; Qu, K. Polystyrene microplastics alter the behavior, energy reserve and nutritional composition of marine jacopever (Sebastes schlegelii). J. Hazard. Mater. 2018, 360, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Ainley, D.G.; Sydeman, W.J.; Parrish, R.H.; Lenarz, W.H. Oceanic factors influencing distribution of young rockfish (Sebastes) in central California—A predators perspective. Calif. Coop. Ocean. Fish. Investag. Rep. 1993, 34, 133–139. [Google Scholar]
- Zhang, Z.; Yu, Y.X.; Jiang, Y.; Wang, Y.G.; Liao, M.J.; Rong, X.J.; Wang, K.; Zhang, H.; Chen, J. First report of isolation and complete genome of Vibrio rotiferianus strain SSVR1601 from cage-cultured black rockfish (Sebastes schlegelii) associated with skin ulcer. J. Fish. Dis. 2019, 42, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Yan, X.; Yang, N.; Fu, Q.; Xue, T.; Zhao, S.; Hu, J.; Li, Q.; Song, L.; Zhang, X.; et al. Genome-wide characterization of Toll-like receptors in black rockfish Sebastes schlegelii: Evolution and response mechanisms following Edwardsiella tarda infection. Int. J. Bio Macromol. 2020, 164, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Han, Z.; Zhang, Z.; Luo, J.; Yanagimoto, T.; Zhang, H. Population genetic differentiation of the black rockfish Sebastes schlegelii revealed by microsatellites. Biochem. Syst. Ecol. 2016, 68, 170–177. [Google Scholar] [CrossRef]
- Niu, J.; Wang, X.; Liu, P.; Liu, H.; Li, R.; Li, Z.; He, Y.; Qi, J. Effects of cryopreservation on sperm with cryodiluent in viviparous Black Rockfish (Sebastes schlegelii). Int. J. Mol. Sci. 2022, 23, 3392. [Google Scholar] [CrossRef]
- Shen, F.; Zhang, Z.; Fu, Y.; Zhang, Z.; Sun, X.; Dong, J.; Ding, X.; Chen, M.; Zhang, X. Effects of food deprivation duration on the behavior and metabolism of black rockfish (Sebastes schlegelii). Fishes 2021, 6, 58. [Google Scholar] [CrossRef]
- Rogers, S.I. Environmental factors affecting the distribution of sole (Solea solea L.) within a nursery area. Neth. J. Sea Res. 1992, 29, 153–161. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Gong, P.; Guan, C. Effects of the artificial reef and flow field environment on the habitat selection behavior of Sebastes schlegelii juveniles. Appl. Anim. Behav. Sci. 2021, 245, 105492. [Google Scholar] [CrossRef]
- Xu, T.; Zhang, X.; Ruan, Z.; Yu, H.; Chen, J.; Jiang, S.; Bian, C.; Wu, B.; Shi, Q.; You, X. Genome resequencing of the orange-spotted grouper (Epinephelus coioides) for a genome-wide association study on ammonia tolerance. Aquaculture 2019, 512, 734332. [Google Scholar] [CrossRef]
- Xu, S.Y.; Zhao, L.L.; Xiao, S.J.; Gao, T.X. Whole genome resequencing data for three rockfish species of Sebastes. Sci. Data 2019, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, T.; Chen, J.; Wu, L.; Wu, X.; Zhang, W.; Luo, J.; Xia, J.; Meng, Z.; Liu, X. Whole-genome sequencing of brown-marbled grouper (Epinephelus fuscoguttatus) provides insights into adaptive evolution and growth differences. Mol. Ecol. Resour. 2022, 22, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yanagimoto, T.; Zhang, X.; Song, N.; Gao, T. Lack of population genetic differentiation of a marine ovoviviparous fish Sebastes schlegelii in Northwestern Pacific. Mitochondrial DNA Part A 2016, 27, 1748–1754. [Google Scholar]
- Zhang, H.; Zhang, Y.; Zhang, X.M.; Song, N.; Gao, T.X. Special structure of mitochondrial DNA control region and phylogenetic relationship among individuals of the black rockfish, Sebastes schlegelii. Mitochondrial DNA 2013, 24, 151–157. [Google Scholar] [CrossRef]
- Hui, Z.; Xiumei, Z.; Zhiqiang, H.; Tianxiang, G. AFLP markers suggest low population genetic differentiation of the black rockfish Sebastes schlegelii. Biochem. Syst. Ecol. 2015, 59, 325–330. [Google Scholar]
- Kramvis, A.; Bukofzer, S.; Kew, M.C. Comparison of hepatitis B virus DNA extractions from serum by the QIAamp blood kit, GeneReleaser, and the phenol-chloroform method. J. Clin. Microbiol. 1996, 34, 2731–2733. [Google Scholar] [CrossRef]
- Han, Z.Q.; Guo, X.Y.; Liu, Q.; Liu, S.S.; Zhang, Z.X.; Xiao, S.J.; Gao, T.-X. Whole-genome resequencing of Japanese whiting (Sillago japonica) provide insights into local adaptations. Zool. Res. 2021, 42, 548–561. [Google Scholar] [CrossRef]
- de Sena, B.G.; Smith, A.D. Falco: High-speed FastQC emulation for quality control of sequencing data. F1000Res 2019, 8, 1874. [Google Scholar] [CrossRef]
- He, Y.; Chang, Y.; Bao, L.; Yu, M.; Li, R.; Niu, J.; Fan, G.; Song, W.; Seim, I.; Qin, Y.; et al. A chromosome-level genome of black rockfish, Sebastes schlegelii, provides insights into the evolution of live birth. Mol. Eco Resour. 2019, 19, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Giannoulatou, E.; Park, S.H.; Humphreys, D.T.; Ho, J.W. Verification and validation of bioinformatics software without a gold standard: A case study of BWA and Bowtie. BMC Bioinform. 2014, 15, S15. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Van-der, A.; Geraldine, A.; Brian, D.O. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E. Visscher PM. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasi, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omcs 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Serrote, C.M.L.; Reiniger, L.R.S.; Silva, K.B.; dos Santos Rabaiolli, S.M.; Stefanel, C.M. Determining the polymorphism information content of a molecular marker. Gene 2020, 726, 144175. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.T.; Ji, Y.; Chang, Y.W.; Shen, Y.; Tian, Z.H.; Gong, W.R.; Du, Y.Z. Population genetic structure and migration patterns of Liriomyza sativae in China: Moderate subdivision and no bridgehead effect revealed by microsatellites. Bull. Entomol. Res. 2016, 106, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Hua, W.; Chen, Y.; Wang, W.; Xue, Z. Comparative analysis of the population diversity of black rockfish (Sebastes schlegelii) in northern China. Mol. Biol. Rep. 2023, 50, 10015–10024. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zhang, M.; Wu, N.; Han, H.; Zhong, R.; Yu, T.; Zheng, Y. Population genetics analysis of the black rockfish Sebastes schlegelii in Northern China based on 2b-RAD simplified genome sequencing. Isr. J. Aquacul.-Bamid 2023, 75, 1–10. [Google Scholar] [CrossRef]
- Orlova, S.Y.; Rastorguev, S.; Bagno, T.; Kurnosov, D.; Nedoluzhko, A. Genetic structure of marine and lake forms of Pacific herring Clupea pallasii. PeerJ 2021, 9, e12444. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, J.; Liu, S.; Song, P.; Guan, Y.; Shan, B.; Lin, L. Genetic diversity of the yellowfin seabream, Acanthopagrus latus (Actinopterygii: Perciformes: Sparidae)—An enhancement species in Dongshan Bay. Acta Ichthyol. Piscat. 2021, 51, 281–287. [Google Scholar] [CrossRef]
- Appleyard, S.A.; Lynch, T.P.; Green, M.A.; Encinas-Viso, F. Genetic diversity and restricted genetic connectivity in an endangered marine fish (Brachionichthys hirsutus) provides a model for conservation management in related and data-deficient species. Mar. Freshw. Res. 2021, 72, 1735–1745. [Google Scholar] [CrossRef]
- Xu, T.; Sun, J.; Watanabe, H.K.; Chen, C.; Nakamura, M.; Ji, R.; Feng, D.; Lv, J.; Wang, S.; Bao, Z.; et al. Population genetic structure of the deep-sea mussel Bathymodiolus platifrons (Bivalvia: Mytilidae) in the Northwest Pacific. Evol. Appl. 2018, 11, 1915–1930. [Google Scholar] [CrossRef]
- Hasson, E.; Eanes, W.F. Contrasting histories of three gene regions associated with in (3L) Payne of Drosophila melanogaster. Genetics 1996, 144, 1565–1575. [Google Scholar] [CrossRef]
- Dobson, A.; Crawley, M. Pathogens and the structure of plant communities. Trends Ecol. Evol. 1994, 9, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Freiwald, J.; Bernardi, G. Genetic diversity affects the strength of population regulation in a marine fish. Ecology 2016, 97, 627–639. [Google Scholar] [CrossRef]
- Li, Y.; Lou, F.R.; Liu, S.G.; Li, H.; Xiang, J.L.; Shan, B.B.; Lin, L.; Zhuang, X. Differentiation and temperature adaptation of Pampus echinogaster based on genome-wide SNPs. Front. Mar. Sci. 2022, 9, 936217. [Google Scholar] [CrossRef]
- Guo, C.; Li, Y.; Xie, J.H.; Han, L.S.; Wang, Y.Q.; Zhang, X.L.; Wu, Y.; Song, J.; Chang, Y.; Ding, J. Revealing selection in breeding and genetic characteristics of economically important traits of new species of Apostichopus japonicas based on genome resequencing and GWAS analysis. Front. Mar. Sci. 2022, 9, 948882. [Google Scholar] [CrossRef]
- Yang, T.Y.; Huang, X.; Jiang, Y.L. Reveal the population genetic characteristics of bombay duck (Harpadon nehereus) in coastal waters of China with Genotype-by-Sequencing technique. J. Ocean. Univ. China 2022, 21, 1373–1380. [Google Scholar] [CrossRef]
- Xu, S.Y.; Yanagimoto, T.; Song, N.; Cai, S.S.; Gao, T.X.; Zhang, X.M. Population genomics reveals possible genetic evidence for parallel evolution of Sebastiscus marmoratus in the northwestern Pacific Ocean. Open Biol. 2019, 9, 190028. [Google Scholar] [CrossRef]
- Chen, Y.L.; Shan, X.J.; Ovando, D.; Yang, T.; Dai, F.Q.; Jin, X.S. Predicting current and future global distribution of black rockfish (Sebastes schlegelii) under changing climate. Ecol. Indic. 2021, 128, 107799. [Google Scholar] [CrossRef]
- Sarvari, P.; Rasouli, S.J.; Allanki, S.; Stone, O.A.; Sokol, A.M.; Graumann, J.; Stainier, D.Y. The E3 ubiquitin-protein ligase Rbx1 regulates cardiac wall morphogenesis in zebrafish. Dev. Biol. 2021, 480, 1–12. [Google Scholar] [CrossRef]
- Mosor, M.; Ziolkowska-Suchanek, I.; Nowicka, K.; Dzikiewicz-Krawczyk, A.; Januszkiewicz-Lewandowska, D.; Nowak, J. Germline variants in MRE11/RAD50/NBN complex genes in childhood leukemia. BMC Cancer 2013, 13, 457. [Google Scholar] [CrossRef]
- Wang, J.; Ji, X.; Liu, J.; Zhang, X. Serine/Threonine Protein Kinase STK16. Int. J. Mol. Sci. 2019, 20, 1760. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhong, B.; Zhao, L.; Hou, Y.; Wang, X.; Chen, X. Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) inhibitors Necrostatin-1 (Nec-1) and 7-Cl-O-Nec-1 (Nec-1s) are potent inhibitors of NAD(P)H: Quinone oxidoreductase 1 (NQO1). Free Radic. Biol. Med. 2021, 173, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Black, S.; Kushner, I.; Samols, D. C-reactive protein. J. Biol. Chem. 2004, 279, 48487–48490. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DL | QD | YT | ||
|---|---|---|---|---|
| Samples | 20 | 20 | 20 | |
| SNPs | 157,778 | 174,480 | 188,756 | |
| Indels | 42,033 | 48,195 | 50,396 | |
| Intergenic | 107,709 (68.27%) | 120,457 (69.04%) | 129,432 (68.57%) | |
| Intron | 88,180 | 98,059 | 105,455 | |
| Exon | 4444 | 4810 | 4948 | |
| Synonymous | 2377 (53.49%) | 2690 (55.93%) | 2662 (53.80%) | |
| Non-synonymous | 2067 (46.51%) | 2120 (44.07%) | 2316 (46.20%) | |
| Splicing | 628 | 644 | 703 | |
| Upstream | 31,285 | 36,050 | 38,776 | |
| Downstream | 31,507 | 35,416 | 37,971 | |
| Population | Observed Heterozygosity (Ho) | Expected Heterozygosity (He) | Polymorphism Information Content (PIC) |
|---|---|---|---|
| DL | 0.14316 | 0.14035 | 0.20672 |
| QD | 0.15809 | 0.15420 | 0.22616 |
| YT | 0.17684 | 0.17145 | 0.24678 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, B.; Gao, T.; He, Y.; Qu, Y.; Zhang, X. Population Genomics of Commercial Fish Sebastes schlegelii of the Bohai and Yellow Seas (China) Using a Large SNP Panel from GBS. Genes 2024, 15, 534. https://doi.org/10.3390/genes15050534
Zhu B, Gao T, He Y, Qu Y, Zhang X. Population Genomics of Commercial Fish Sebastes schlegelii of the Bohai and Yellow Seas (China) Using a Large SNP Panel from GBS. Genes. 2024; 15(5):534. https://doi.org/10.3390/genes15050534
Chicago/Turabian StyleZhu, Beiyan, Tianxiang Gao, Yan He, Yinquan Qu, and Xiumei Zhang. 2024. "Population Genomics of Commercial Fish Sebastes schlegelii of the Bohai and Yellow Seas (China) Using a Large SNP Panel from GBS" Genes 15, no. 5: 534. https://doi.org/10.3390/genes15050534
APA StyleZhu, B., Gao, T., He, Y., Qu, Y., & Zhang, X. (2024). Population Genomics of Commercial Fish Sebastes schlegelii of the Bohai and Yellow Seas (China) Using a Large SNP Panel from GBS. Genes, 15(5), 534. https://doi.org/10.3390/genes15050534