
A Novel COL4A5 Pathogenic Variant Joins the Dots in a Family with a Synchronous Diagnosis of Alport Syndrome and Polycystic Kidney Disease

 , , , , and
, , , , and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
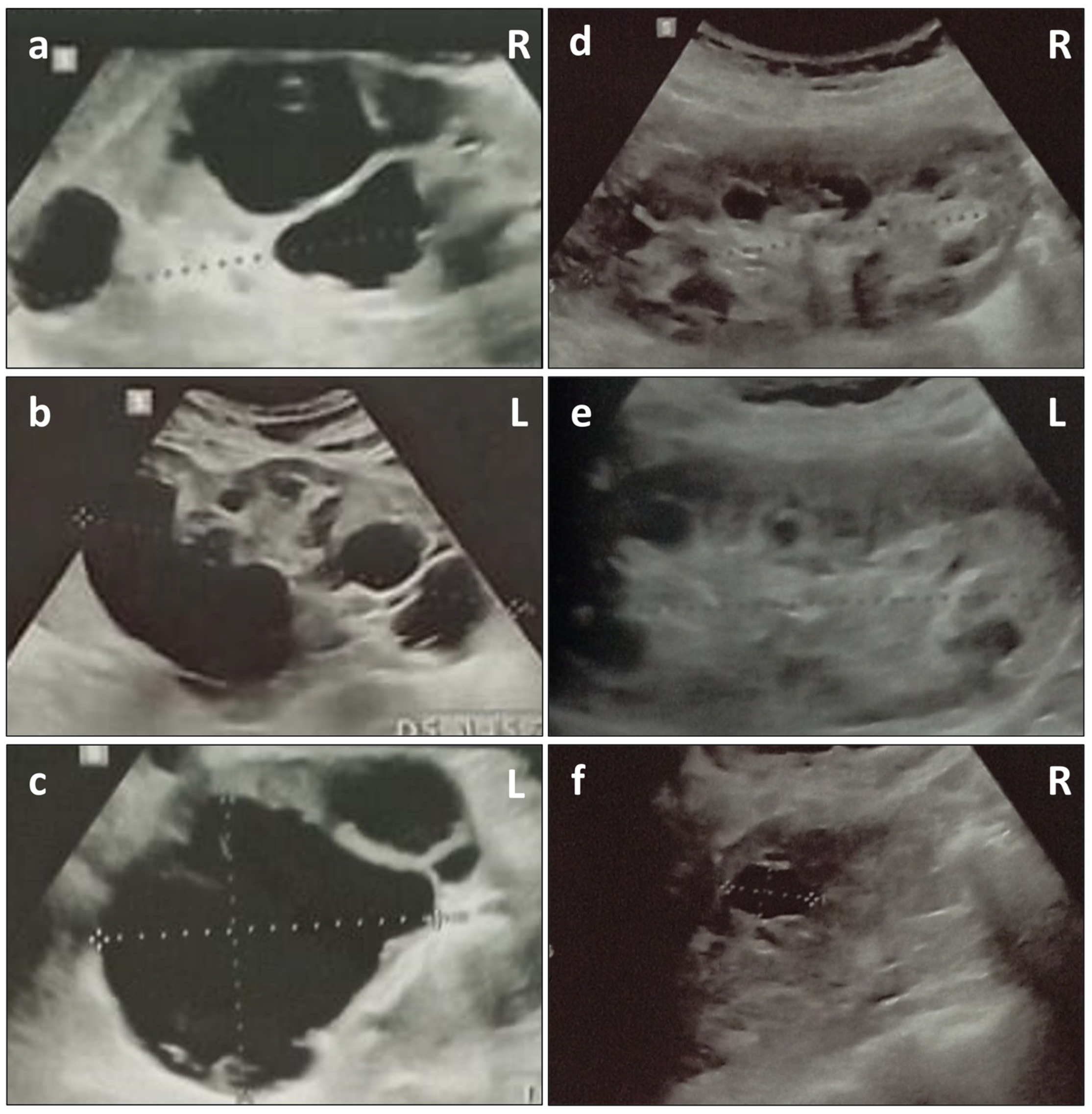
2. Case Presentation
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warady, B.A.; Agarwal, R.; Bangalore, S.; Chapman, A.; Levin, A.; Stenvinkel, P.; Toto, R.D.; Chertow, G.M. Alport Syndrome Classification and Management. Kidney Med. 2020, 2, 639–649. [Google Scholar] [CrossRef]
- Watson, S.; Padala, S.A.; Hashmi, M.F.; Bush, J.S. Alport Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK470419/ (accessed on 21 April 2024).
- Savige, J.; Storey, H.; Watson, E.; Hertz, J.M.; Deltas, C.; Renieri, A.; Mari, F.; Hilbert, P.; Plevova, P.; Byers, P.; et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: Refining the ACMG criteria. Eur. J. Hum. Genet. 2021, 29, 1186–1197. [Google Scholar] [CrossRef]
- Savige, J.; Renieri, A.; Ars, E.; Daga, S.; Pinto, A.M.; Rothe, H.; Gale, D.P.; Aksenova, M.; Cerkauskaite, A.; Bielska, O.; et al. Digenic Alport Syndrome. Clin. J. Am. Soc. Nephrol. CJASN 2022, 17, 1697–1706. [Google Scholar] [CrossRef]
- Daga, S.; Ding, J.; Deltas, C.; Savige, J.; Lipska-Ziętkiewicz, B.S.; Hoefele, J.; Flinter, F.; Gale, D.P.; Aksenova, M.; Kai, H.; et al. The 2019 and 2021 International Workshops on Alport Syndrome. Eur. J. Hum. Genet. 2022, 30, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.; Fieldhouse, R.; Chan, M.M.Y.; Sadeghi-Alavijeh, O.; Burnett, L.; Izzi, V.; Persikov, A.V.; Gale, D.P.; Storey, H.; Savige, J.; et al. Prevalence Estimates of Predicted Pathogenic COL4A3–COL4A5 Variants in a Population Sequencing Database and Their Implications for Alport Syndrome. J. Am. Soc. Nephrol. 2021, 32, 2273–2290. [Google Scholar] [CrossRef]
- Boudko, S.P.; Danylevych, N.; Hudson, B.G.; Pedchenko, V.K. Basement membrane collagen IV: Isolation of functional domains. In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 143, pp. 171–185. ISBN 978-0-12-812297-6. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0091679X17301310 (accessed on 25 January 2024).
- Heidet, L.; Cai, Y.; Guicharnaud, L.; Antignac, C.; Gubler, M.-C. Glomerular Expression of Type IV Collagen Chains in Normal and X-Linked Alport Syndrome Kidneys. Am. J. Pathol. 2000, 156, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Jais, J.P.; Knebelmann, B.; Giatras, I.; Marchi, M.D.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.-O.; Flinter, F.; et al. X-linked Alport Syndrome: Natural History in 195 Families and Genotype- Phenotype Correlations in Males. J. Am. Soc. Nephrol. 2000, 11, 649–657. [Google Scholar] [CrossRef]
- Yuan, X.; Su, Q.; Wang, H.; Shi, S.; Liu, L.; Lv, J.; Wang, S.; Zhu, L.; Zhang, H. Genetic Variants of the COL4A3, COL4A4, and COL4A5 Genes Contribute to Thinned Glomerular Basement Membrane Lesions in Sporadic IgA Nephropathy Patients. J. Am. Soc. Nephrol. JASN 2023, 34, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Yavaş, C.; Ozdemir Ozgenturk, N.; Dogan, M.; Gezdirici, A.; Keskin, E.; Gokpınar İli, E.; Dogan, T.; Celebi, E.; Bender, O.; Un, C. A Deeper Insight into COL4A3, COL4A4, and COL4A5 Variants and Genotype-Phenotype Correlation of a Turkish Cohort with Alport Syndrome. Mol. Syndromol. 2024, 15, 1–13. [Google Scholar] [CrossRef]
- Gast, C.; Pengelly, R.J.; Lyon, M.; Bunyan, D.J.; Seaby, E.G.; Graham, N.; Venkat-Raman, G.; Ennis, S. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2016, 31, 961–970. [Google Scholar] [CrossRef]
- Savige, J.; Mack, H.; Thomas, R.; Langsford, D.; Pianta, T. Alport Syndrome with Kidney Cysts Is Still Alport Syndrome. Kidney Int. Rep. 2022, 7, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Gulati, A.; Sevillano, A.M.; Praga, M.; Gutierrez, E.; Alba, I.; Dahl, N.K.; Besse, W.; Choi, J.; Somlo, S. Collagen IV Gene Mutations in Adults with Bilateral Renal Cysts and CKD. Kidney Int. Rep. 2020, 5, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Zeni, L.; Mescia, F.; Toso, D.; Dordoni, C.; Mazza, C.; Savoldi, G.; Econimo, L.; Cortinovis, R.; Fisogni, S.; Alberici, F.; et al. Clinical Significance of the Cystic Phenotype in Alport Syndrome. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2024. [Google Scholar] [CrossRef]
- Furlano, M.; Pilco-Teran, M.; Pybus, M.; Martínez, V.; Aza-Carmona, M.; Rius, A.; Pérez-Gomez, V.; Berná, G.; Mazo, J.; Hernández, J.; et al. Increased prevalence of Kidney cysts in individuals carrying heterozygous COL4A3 or COL4A4 pathogenic variants. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2024, gfae031. [Google Scholar] [CrossRef] [PubMed]
- Bada-Bosch, T.; Sevillano, A.M.; Teresa Sánchez-Calvin, M.; Palma-Milla, C.; Alba de Cáceres, I.; Díaz-Crespo, F.; Trujillo, H.; Alonso, M.; Cases-Corona, C.; Shabaka, A.; et al. Cystic phenotype and chronic kidney disease in autosomal dominant Alport syndrome. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2024, gfae002. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, S.R.; Aeddula, N.R. Chronic Kidney Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK535404/ (accessed on 9 March 2024).
- Inker, L.A.; Eneanya, N.D.; Coresh, J.; Tighiouart, H.; Wang, D.; Sang, Y.; Crews, D.C.; Doria, A.; Estrella, M.M.; Froissart, M.; et al. New Creatinine- and Cystatin C-Based Equations to Estimate GFR without Race. N. Engl. J. Med. 2021, 385, 1737–1749. [Google Scholar] [CrossRef] [PubMed]
- Silverman, S.G.; Pedrosa, I.; Ellis, J.H.; Hindman, N.M.; Schieda, N.; Smith, A.D.; Remer, E.M.; Shinagare, A.B.; Curci, N.E.; Raman, S.S.; et al. Bosniak Classification of Cystic Renal Masses, Version 2019: An Update Proposal and Needs Assessment. Radiology 2019, 292, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Torres, V.E.; Harris, P.C. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J. Am. Soc. Nephrol. 2018, 29, 13–23. [Google Scholar] [CrossRef]
- Gettelfinger, J.D.; Dahl, J.P. Syndromic Hearing Loss: A Brief Review of Common Presentations and Genetics. J. Pediatr. Genet. 2018, 7, 1–8. [Google Scholar] [CrossRef]
- Savige, J.; Storey, H.; Il Cheong, H.; Gyung Kang, H.; Park, E.; Hilbert, P.; Persikov, A.; Torres-Fernandez, C.; Ars, E.; Torra, R.; et al. X-Linked and Autosomal Recessive Alport Syndrome: Pathogenic Variant Features and Further Genotype-Phenotype Correlations. PLoS ONE 2016, 11, e0161802. [Google Scholar] [CrossRef] [PubMed]
- Bekheirnia, M.R.; Reed, B.; Gregory, M.C.; McFann, K.; Shamshirsaz, A.A.; Masoumi, A.; Schrier, R.W. Genotype–Phenotype Correlation in X-Linked Alport Syndrome. J. Am. Soc. Nephrol. 2010, 21, 876–883. [Google Scholar] [CrossRef]
- Rheault, M.N. Women and Alport syndrome. Pediatr. Nephrol. 2012, 27, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Chew, C.; Lennon, R. Basement Membrane Defects in Genetic Kidney Diseases. Front. Pediatr. 2018, 6, 11. [Google Scholar] [CrossRef]
- Cosgrove, D.; Madison, J. Molecular and Cellular Mechanisms Underlying the Initiation and Progression of Alport Glomerular Pathology. Front. Med. 2022, 9, 846152. [Google Scholar] [CrossRef]
- Savige, J.; Harraka, P. Pathogenic Variants in the Genes Affected in Alport Syndrome (COL4A3–COL4A5) and Their Association with Other Kidney Conditions: A Review. Am. J. Kidney Dis. 2021, 78, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Rahbari-Oskoui, F.; O’Neill, W.C. Diagnosis and Management of Acquired Cystic Kidney Disease and Renal Tumors in ESRD Patients. Semin. Dial. 2017, 30, 373–379. [Google Scholar] [CrossRef]
- Ishikawa, I. Acquired cystic disease: Mechanisms and manifestations. Semin. Nephrol. 1991, 11, 671–684. [Google Scholar]
- Grantham, J.J. Acquired cystic kidney disease. Kidney Int. 1991, 40, 143–152. [Google Scholar] [CrossRef]
- Hood, J.C. Correlation of histopathological features and renal impairment in autosomal dominant Alport syndrome in Bull terriers. Nephrol. Dial. Transplant. 2002, 17, 1897–1908. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Migeon, T.; Verpont, M.-C.; Zaidan, M.; Sado, Y.; Kerjaschki, D.; Ronco, P.; Plaisier, E. HANAC Syndrome Col4a1 Mutation Causes Neonate Glomerular Hyperpermeability and Adult Glomerulocystic Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Plaisier, E.; Gribouval, O.; Alamowitch, S.; Mougenot, B.; Prost, C.; Verpont, M.C.; Marro, B.; Desmettre, T.; Cohen, S.Y.; Roullet, E.; et al. COL4A1 Mutations and Hereditary Angiopathy, Nephropathy, Aneurysms, and Muscle Cramps. N. Engl. J. Med. 2007, 357, 2687–2695. [Google Scholar] [CrossRef] [PubMed]
- Hanna, C.; Iliuta, I.-A.; Besse, W.; Mekahli, D.; Chebib, F.T. Cystic Kidney Diseases in Children and Adults: Differences and Gaps in Clinical Management. Semin. Nephrol. 2023, 43, 151434. [Google Scholar] [CrossRef] [PubMed]
- Ravine, D.; Gibson, R.N.; Donlan, J.; Sheffield, L.J. An Ultrasound Renal Cyst Prevalence Survey: Specificity Data for Inherited Renal Cystic Diseases. Am. J. Kidney Dis. 1993, 22, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.B.; Devuyst, O.; Eckardt, K.-U.; Gansevoort, R.T.; Harris, T.; Horie, S.; Kasiske, B.L.; Odland, D.; Pei, Y.; Perrone, R.D.; et al. Autosomal-dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88, 17–27. [Google Scholar] [CrossRef]
- Capuano, I.; Buonanno, P.; Riccio, E.; Rizzo, M.; Pisani, A. Tolvaptan vs. somatostatin in the treatment of ADPKD: A review of the literature. Clin. Nephrol. 2022, 97, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Urate, S.; Yamada, T.; Azushima, K.; Yamaji, T.; Kinguchi, S.; Uneda, K.; Kanaoka, T.; Wakui, H.; Tamura, K. Comparative Efficacy of Pharmacological Treatments for Adults with Autosomal Dominant Polycystic Kidney Disease: A Systematic Review and Network Meta-Analysis of Randomized Controlled Trials. Front. Pharmacol. 2022, 13, 885457. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Licht, C.; Anders, H.J.; Hoppe, B.; Beck, B.; Tönshoff, B.; Höcker, B.; Wygoda, S.; Ehrich, J.H.H.; Pape, L.; et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012, 81, 494–501. [Google Scholar] [CrossRef]
- Temme, J.; Peters, F.; Lange, K.; Pirson, Y.; Heidet, L.; Torra, R.; Grunfeld, J.-P.; Weber, M.; Licht, C.; Müller, G.-A.; et al. Incidence of renal failure and nephroprotection by RAAS inhibition in heterozygous carriers of X-chromosomal and autosomal recessive Alport mutations. Kidney Int. 2012, 81, 779–783. [Google Scholar] [CrossRef]
- Kashtan, C.E.; Ding, J.; Gregory, M.; Gross, O.; Heidet, L.; Knebelmann, B.; Rheault, M.; Licht, C. Clinical practice recommendations for the treatment of Alport syndrome: A statement of the Alport Syndrome Research Collaborative. Pediatr. Nephrol. 2013, 28, 5–11. [Google Scholar] [CrossRef]
- Luciano, R.L.; Dahl, N.K. Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): Considerations for routine screening and management. Nephrol. Dial. Transplant. 2014, 29, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Obeidova, L.; Seeman, T.; Fencl, F.; Blahova, K.; Hojny, J.; Elisakova, V.; Reiterova, J.; Stekrova, J. Results of targeted next-generation sequencing in children with cystic kidney diseases often change the clinical diagnosis. PLoS ONE 2020, 15, e0235071. [Google Scholar] [CrossRef] [PubMed]
- Groopman, E.E.; Marasa, M.; Cameron-Christie, S.; Petrovski, S.; Aggarwal, V.S.; Milo-Rasouly, H.; Li, Y.; Zhang, J.; Nestor, J.; Krithivasan, P.; et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N. Engl. J. Med. 2019, 380, 142–151. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graziani, L.; Minotti, C.; Carriero, M.L.; Bengala, M.; Lai, S.; Terracciano, A.; Novelli, A.; Novelli, G. A Novel COL4A5 Pathogenic Variant Joins the Dots in a Family with a Synchronous Diagnosis of Alport Syndrome and Polycystic Kidney Disease. Genes 2024, 15, 597. https://doi.org/10.3390/genes15050597
Graziani L, Minotti C, Carriero ML, Bengala M, Lai S, Terracciano A, Novelli A, Novelli G. A Novel COL4A5 Pathogenic Variant Joins the Dots in a Family with a Synchronous Diagnosis of Alport Syndrome and Polycystic Kidney Disease. Genes. 2024; 15(5):597. https://doi.org/10.3390/genes15050597
Chicago/Turabian StyleGraziani, Ludovico, Chiara Minotti, Miriam Lucia Carriero, Mario Bengala, Silvia Lai, Alessandra Terracciano, Antonio Novelli, and Giuseppe Novelli. 2024. "A Novel COL4A5 Pathogenic Variant Joins the Dots in a Family with a Synchronous Diagnosis of Alport Syndrome and Polycystic Kidney Disease" Genes 15, no. 5: 597. https://doi.org/10.3390/genes15050597