
Inherited Retinal Diseases and Retinal Organoids as Preclinical Cell Models for Inherited Retinal Disease Research

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Human Retina
2.1. Structure and Function of the Human Retina
2.2. Development of the Retina
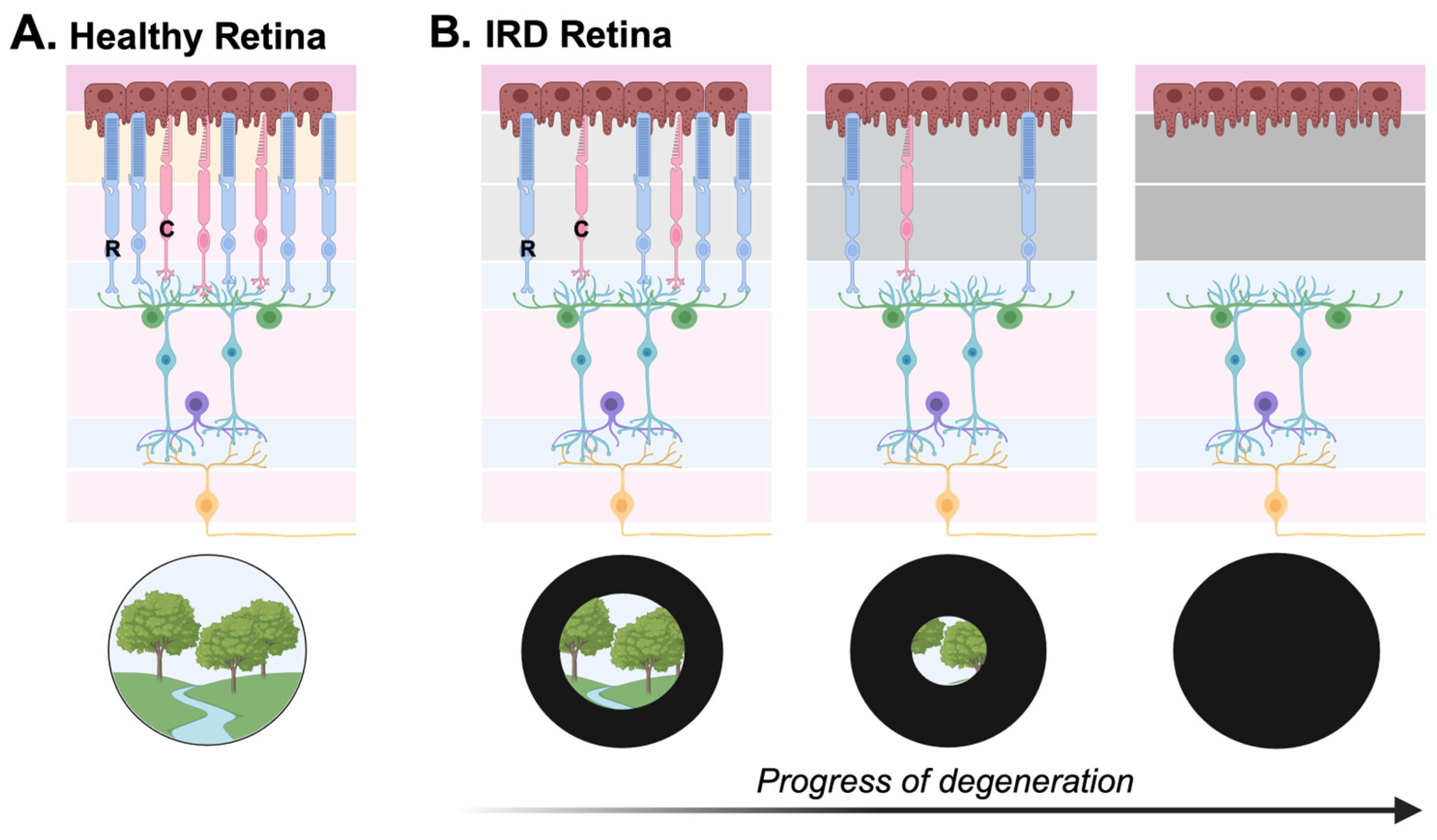
3. Inherited Retinal Diseases
3.1. Genetics of Inherited Retinal Diseases
3.2. Clinical Classifications of Inherited Retinal Diseases
4. Current Approaches to IRD Treatment Discovery
4.1. Gene Therapy
4.2. Cell Therapy
4.3. Models for IRD Research
5. Retinal Organoids as a Preclinical Cell Model
5.1. The Origin of Retinal Organoids
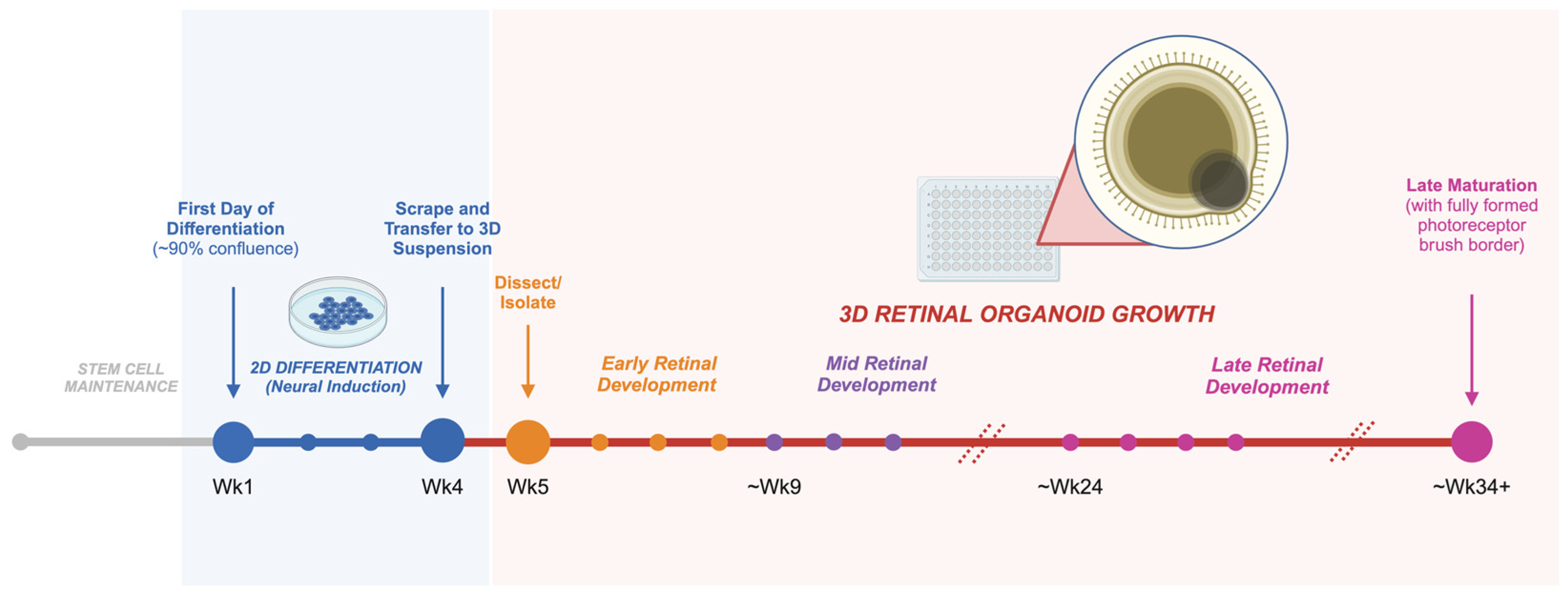
5.2. Growing Retinal Organoids from hPSCs
5.3. Recapitulating Human Retinal Development with Retinal Organoids
5.4. Use of Retinal Organoids in IRD Research
5.4.1. Retinal Organoids for the Discovery of Novel IRD Variants
5.4.2. Retinal Organoids to Study Variant-Specific IRD Pathogenesis and Disease Mechanisms
5.4.3. Retinal Organoids for the Discovery of IRD Therapies
5.5. Limitations of Retinal Organoids as a Preclinical Model
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Martinez Velazquez, L.A.; Ballios, B.G. The Next Generation of Molecular and Cellular Therapeutics for Inherited Retinal Disease. Int. J. Mol. Sci. 2021, 22, 11542. [Google Scholar] [CrossRef]
- Dowling, J.E. The Retina: An Approachable Part of the Brain; Rev. ed.; Belknap Press of Harvard University Press: Cambridge, MA, USA, 2012; ISBN 978-0-674-06154-5. [Google Scholar]
- Quinn, P.M.J.; Wijnholds, J. Retinogenesis of the Human Fetal Retina: An Apical Polarity Perspective. Genes 2019, 10, 987. [Google Scholar] [CrossRef]
- Marquardt, T.; Gruss, P. Generating Neuronal Diversity in the Retina: One for Nearly All. Trends Neurosci. 2002, 25, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Cepko, C.L.; Austin, C.P.; Yang, X.; Alexiades, M.; Ezzeddine, D. Cell Fate Determination in the Vertebrate Retina. Proc. Natl. Acad. Sci. USA 1996, 93, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, A.; Djajadi, H.; Erickson, A.; Possin, D. Development of the Human Retina in the Absence of Ganglion Cells. Exp. Eye Res. 2006, 83, 920–931. [Google Scholar] [CrossRef]
- Ben-Yosef, T. Inherited Retinal Diseases. Int. J. Mol. Sci. 2022, 23, 13467. [Google Scholar] [CrossRef]
- Aziz, K.; Swenor, B.K.; Canner, J.K.; Singh, M.S. The Direct Healthcare Cost of Stargardt Disease: A Claims-Based Analysis. Ophthalmic Epidemiol. 2021, 28, 533–539. [Google Scholar] [CrossRef]
- Schneider, N.; Sundaresan, Y.; Gopalakrishnan, P.; Beryozkin, A.; Hanany, M.; Levanon, E.Y.; Banin, E.; Ben-Aroya, S.; Sharon, D. Inherited Retinal Diseases: Linking Genes, Disease-Causing Variants, and Relevant Therapeutic Modalities. Prog. Retin. Eye Res. 2022, 89, 101029. [Google Scholar] [CrossRef]
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [PubMed]
- Dhurandhar, D.; Sahoo, N.; Mariappan, I.; Narayanan, R. Gene Therapy in Retinal Diseases: A Review. Indian J. Ophthalmol. 2021, 69, 2257. [Google Scholar] [CrossRef]
- Amato, A.; Arrigo, A.; Aragona, E.; Manitto, M.P.; Saladino, A.; Bandello, F.; Battaglia Parodi, M. Gene Therapy in Inherited Retinal Diseases: An Update on Current State of the Art. Front. Med. 2021, 8, 750586. [Google Scholar] [CrossRef] [PubMed]
- Pierce, E.A.; Aleman, T.S.; Jayasundera, K.T.; Ashimatey, B.S.; Kim, K.; Rashid, A.; Jaskolka, M.C.; Myers, R.L.; Lam, B.L.; Bailey, S.T.; et al. Gene Editing for CEP290-Associated Retinal Degeneration. N. Engl. J. Med. 2024; advance online publication. [Google Scholar] [CrossRef]
- ProQR Therapeutics. An Open-Label, Dose Escalation and Double-Masked, Randomized, Controlled Trial Evaluating Safety and Tolerability of Sepofarsen in Children (<8 Years of Age) with LCA10 Caused by Mutations in the CEP290 Gene. ClinicalTrials.gov Identifier: NCT04855045. Updated 25 March 2022. Available online: https://www.clinicaltrials.gov/study/NCT04855045 (accessed on 8 March 2024).
- Russell, S.R.; Drack, A.V.; Cideciyan, A.V.; Jacobson, S.G.; Leroy, B.P.; Van Cauwenbergh, C.; Ho, A.C.; Dumitrescu, A.V.; Han, I.C.; Martin, M.; et al. Intravitreal antisense oligonucleotide sepofarsen in Leber congenital amaurosis type 10: A phase 1b/2 trial. Nat. Med. 2022, 28, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- ProQR Therapeutics. Study to Evaluate Safety and Tolerability of QR-421a in Subjects with RP Due to Mutations in Exon 13 of the USH2A Gene. ClinicalTrials.gov Identifier: NCT03780257. Updated 20 April 2022. Available online: https://www.clinicaltrials.gov/study/NCT03780257 (accessed on 8 March 2024).
- Dulla, K.; Slijkerman, R.; van Diepen, H.C.; Albert, S.; Dona, M.; Beumer, W.; Turunen, J.J.; Chan, H.L.; Schulkens, I.A.; Vorthoren, L.; et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol. Ther. 2021, 29, 2441–2455. [Google Scholar] [CrossRef]
- Nanoscope Therapeutics Inc. Efficacy and Safety of MCO-010 Optogenetic Therapy in Adults with Retinitis Pigmentosa [RESTORE]. ClinicalTrials.gov Identifier: NCT04945772. Updated 17 January 2024. Available online: https://clinicaltrials.gov/study/NCT04945772 (accessed on 8 March 2024).
- Bionic Sight LLC. BS01 in Patients with Retinitis Pigmentosa. ClinicalTrials.gov Identifier: NCT04278131. Updated 3 May 2023. Available online: https://www.clinicaltrials.gov/study/NCT04278131 (accessed on 8 March 2024).
- GenSight Biologics. Dose-Escalation Study to Evaluate the Safety and Tolerability of GS030 in Subjects with Retinitis Pigmentosa. ClinicalTrials.gov Identifier: NCT03326336. Updated 26 July 2022. Available online: https://www.clinicaltrials.gov/study/NCT03326336 (accessed on 8 March 2024).
- Battu, R.; Ratra, D.; Gopal, L. Newer Therapeutic Options for Inherited Retinal Diseases: Gene and Cell Replacement Therapy. Indian J. Ophthalmol. 2022, 70, 2316. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.; Choi, E.H.; Leinonen, H.; Foik, A.T.; Newby, G.A.; Yeh, W.-H.; Dong, Z.; Kiser, P.D.; Lyon, D.C.; Liu, D.R.; et al. Restoration of Visual Function in Adult Mice with an Inherited Retinal Disease via Adenine Base Editing. Nat. Biomed. Eng. 2020, 5, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Vats, A.; Sahel, J.-A.; Chen, Y.; Byrne, L.C. Gene Augmentation Prevents Retinal Degeneration in a CRISPR/Cas9-Based Mouse Model of PRPF31 Retinitis Pigmentosa. Nat. Commun. 2022, 13, 7695. [Google Scholar] [CrossRef]
- O’Hara-Wright, M.; Gonzalez-Cordero, A. Retinal Organoids: A Window into Human Retinal Development. Development 2020, 147, dev189746. [Google Scholar] [CrossRef]
- Afanasyeva, T.A.V.; Corral-Serrano, J.C.; Garanto, A.; Roepman, R.; Cheetham, M.E.; Collin, R.W.J. A Look into Retinal Organoids: Methods, Analytical Techniques, and Applications. Cell. Mol. Life Sci. 2021, 78, 6505–6532. [Google Scholar] [CrossRef] [PubMed]
- Toualbi, L.; Toms, M.; Moosajee, M. USH2A-Retinopathy: From Genetics to Therapeutics. Exp. Eye Res. 2020, 201, 108330. [Google Scholar] [CrossRef]
- Moshiri, A. Animals Models of Inherited Retinal Disease. Int. Ophthalmol. Clin. 2021, 61, 113–130. [Google Scholar] [CrossRef]
- Perkins, B.D. Zebrafish models of inherited retinal dystrophies. J. Transl. Genet. Genom. 2022, 6, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Osakada, F.; Ikeda, H.; Sasai, Y.; Takahashi, M. Stepwise Differentiation of Pluripotent Stem Cells into Retinal Cells. Nat. Protoc. 2009, 4, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.S.; Shearer, R.L.; Capowski, E.E.; Wright, L.S.; Wallace, K.A.; McMillan, E.L.; Zhang, S.-C.; Gamm, D.M. Modeling Early Retinal Development with Human Embryonic and Induced Pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 16698–16703. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, R.; Ishikawa, Y.; Sasamoto, Y.; Katori, R.; Nomura, N.; Ichikawa, T.; Araki, S.; Soma, T.; Kawasaki, S.; Sekiguchi, K.; et al. Co-ordinated ocular development from human iPS cells and recovery of corneal function. Nature 2016, 531, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Hallam, D.; Hilgen, G.; Dorgau, B.; Zhu, L.; Yu, M.; Bojic, S.; Hewitt, P.; Schmitt, M.; Uteng, M.; Kustermann, S.; et al. Human-Induced Pluripotent Stem Cells Generate Light Responsive Retinal Organoids with Variable and Nutrient-Dependent Efficiency. Stem Cells 2018, 36, 1535–1551. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-Formation of Optic Cups and Storable Stratified Neural Retina from Human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.-H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of Three-Dimensional Retinal Tissue with Functional Photoreceptors from Human IPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed]
- Reichman, S.; Terray, A.; Slembrouck, A.; Nanteau, C.; Orieux, G.; Habeler, W.; Nandrot, E.F.; Sahel, J.-A.; Monville, C.; Goureau, O. From Confluent Human IPS Cells to Self-Forming Neural Retina and Retinal Pigmented Epithelium. Proc. Natl. Acad. Sci. USA 2014, 111, 8518–8523. [Google Scholar] [CrossRef]
- Gonzalez-Cordero, A.; Kruczek, K.; Naeem, A.; Fernando, M.; Kloc, M.; Ribeiro, J.; Goh, D.; Duran, Y.; Blackford, S.J.I.; Abelleira-Hervas, L.; et al. Recapitulation of Human Retinal Development from Human Pluripotent Stem Cells Generates Transplantable Populations of Cone Photoreceptors. Stem Cell Rep. 2017, 9, 820–837. [Google Scholar] [CrossRef]
- Rashidi, H.; Leong, Y.C.; Venner, K.; Pramod, H.; Fei, Q.-Z.; Jones, O.J.R.; Moulding, D.; Sowden, J.C. Generation of 3D Retinal Tissue from Human Pluripotent Stem Cells Using a Directed Small Molecule-Based Serum-Free Microwell Platform. Sci. Rep. 2022, 12, 6646. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, P.; Ma, J.H.; Cui, Z.; Yu, Q.; Liu, S.; Xue, Y.; Zhu, D.; Cao, J.; Li, Z.; et al. Modeling Retinitis Pigmentosa: Retinal Organoids Generated from the IPSCs of a Patient with the USH2A Mutation Show Early Developmental Abnormalities. Front. Cell. Neurosci. 2019, 13, 361. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Lu, Q.; Insinna-Kettenhofen, C.; Nagashima, K.; English, M.A.; Semler, E.M.; Mahgerefteh, J.; Cideciyan, A.V.; Li, T.; Brooks, B.P.; et al. In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations. Cell Rep. 2017, 20, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.-L.; Lei, X.-L.; Han, F.; He, K.-W.; Jin, S.-Q.; Zhang, Y.-Y.; Jin, Z.-B. Patient-Specific Retinal Organoids Recapitulate Disease Features of Late-Onset Retinitis Pigmentosa. Front. Cell Dev. Biol. 2020, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Boon, N.; Lu, X.; Andriessen, C.A.; Moustakas, I.; Buck, T.M.; Freund, C.; Arendzen, C.H.; Böhringer, S.; Boon, C.J.F.; Mei, H.; et al. AAV-Mediated Gene Augmentation Therapy of CRB1 Patient-Derived Retinal Organoids Restores the Histological and Transcriptional Retinal Phenotype. Stem Cell Rep. 2023, 18, 1123–1137. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo-Soriano, C.; Jimenez-Medina, C.; Erkilic, N.; Cappellino, L.; Lefevre, A.; Nagel-Wolfrum, K.; Wolfrum, U.; Van Wijk, E.; Roux, A.F.; Meunier, I.; et al. USH2A variants causing retinitis pigmentosa or Usher syndrome provoke differential retinal phenotypes in disease-specific organoids. HGG Adv. 2023, 4, 100229. [Google Scholar] [CrossRef] [PubMed]
- Dona, M.; Slijkerman, R.; Lerner, K.; Broekman, S.; Wegner, J.; Howat, T.; Peters, T.; Hetterschijt, L.; Boon, N.; de Vrieze, E.; et al. Usherin defects lead to early-onset retinal dysfunction in zebrafish. Exp. Eye Res. 2018, 173, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Thompson, J.A.; Zhang, D.; Charng, J.; Arunachalam, S.; McLaren, T.L.; Lamey, T.M.; De Roach, J.N.; Jennings, L.; McLenachan, S.; et al. Characterization of CRB1 splicing in retinal organoids derived from a patient with adult-onset rod-cone dystrophy caused by the c.1892A>G and c.2548G>A variants. Mol. Genet. Genom. Med. 2020, 8, e1489. [Google Scholar] [CrossRef] [PubMed]
- Sladen, P.E.; Naeem, A.; Adefila-Ideozu, T.; Vermeule, T.; Busson, S.L.; Michaelides, M.; Naylor, S.; Forbes, A.; Lane, A.; Georgiadis, A. AAV-RPGR Gene Therapy Rescues Opsin Mislocalisation in a Human Retinal Organoid Model of RPGR-Associated X-Linked Retinitis Pigmentosa. Int. J. Mol. Sci. 2024, 25, 1839. [Google Scholar] [CrossRef]
- Vielle, A.; Park, Y.K.; Secora, C.; Vergara, M.N. Organoids for the Study of Retinal Development and Developmental Abnormalities. Front. Cell. Neurosci. 2021, 15, 667880. [Google Scholar] [CrossRef]
- Meyer, J.S.; Howden, S.E.; Wallace, K.A.; Verhoeven, A.D.; Wright, L.S.; Capowski, E.E.; Pinilla, I.; Martin, J.M.; Tian, S.; Stewart, R.; et al. Optic Vesicle-like Structures Derived from Human Pluripotent Stem Cells Facilitate a Customized Approach to Retinal Disease Treatment. Stem Cells 2011, 29, 1206–1218. [Google Scholar] [CrossRef]
- Mao, X.; An, Q.; Xi, H.; Yang, X.-J.; Zhang, X.; Yuan, S.; Wang, J.; Hu, Y.; Liu, Q.; Fan, G. Single-Cell RNA Sequencing of HESC-Derived 3D Retinal Organoids Reveals Novel Genes Regulating RPC Commitment in Early Human Retinogenesis. Stem Cell Rep. 2019, 13, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Dryja, T.P.; McGee, T.L.; Hahn, L.B.; Cowley, G.S.; Olsson, J.E.; Reichel, E.; Sandberg, M.A.; Berson, E.L. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N. Engl. J. Med. 1990, 323, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Cremers, F.P.; van de Pol, D.J.; van Kerkhoff, L.P.; Wieringa, B.; Ropers, H.H. Cloning of a gene that is rearranged in patients with choroideraemia. Nature 1990, 347, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, M.; Duignan, E.; Malone, C.P.; Stephenson, K.; Saad, T.; McDermott, C.; Green, A.; Keegan, D.; Humphries, P.; Kenna, P.F.; et al. Panel-Based Population Next-Generation Sequencing for Inherited Retinal Degenerations. Sci. Rep. 2016, 6, 33248. [Google Scholar] [CrossRef] [PubMed]
- Mullin, N.K.; Voigt, A.P.; Cooke, J.A.; Bohrer, L.R.; Burnight, E.R.; Stone, E.M.; Mullins, R.F.; Tucker, B.A. Patient derived stem cells for discovery and validation of novel pathogenic variants in inherited retinal disease. Prog. Retin. Eye Res. 2021, 83, 100918. [Google Scholar] [CrossRef]
- Bronstein, R.; Capowski, E.E.; Mehrotra, S.; Jansen, A.D.; Navarro-Gomez, D.; Maher, M.; Place, E.; Sangermano, R.; Bujakowska, K.M.; Gamm, D.M.; et al. A combined RNA-seq and whole genome sequencing approach for identification of non-coding pathogenic variants in single families. Hum. Mol. Genet. 2020, 29, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Burnight, E.R.; Fenner, B.J.; Han, I.C.; DeLuca, A.P.; Whitmore, S.S.; Bohrer, L.R.; Andorf, J.L.; Sohn, E.H.; Mullins, R.F.; Tucker, B.A.; et al. Demonstration of the pathogenicity of a common non-exomic mutation in ABCA4 using iPSC-derived retinal organoids and retrospective clinical data. Hum. Mol. Genet. 2023, ddad176. [Google Scholar] [CrossRef]
- Liu, X.; Bulgakov, O.V.; Darrow, K.N.; Pawlyk, B.; Adamian, M.; Liberman, M.C.; Li, T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4413–4418. [Google Scholar] [CrossRef]
- Na, H.J.; Kwon, J.E.; Kim, S.H.; Ahn, J.; Kwon, O.S.; Chung, K.S. Human Pluripotent Stem Cell-Derived Retinal Organoids: A Viable Platform for Investigating the Efficacy of Adeno-Associated Virus Gene Therapy. Int. J. Stem Cells. 2024. [Google Scholar] [CrossRef]
- Deng, W.L.; Gao, M.L.; Lei, X.L.; Lv, J.N.; Zhao, H.; He, K.W.; Xia, X.X.; Li, L.Y.; Chen, Y.C.; Li, Y.P.; et al. Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients. Stem Cell Rep. 2018, 10, 1267–1281. [Google Scholar] [CrossRef]
- Janssen Research & Development, LLC. Gene Therapy Trial for the Treatment of X-Linked Retinitis Pigmentosa Associated with Variants in the RPGR Gene. ClinicalTrials.gov Identifier: NCT04671433. Updated 28 February 2024. Available online: https://clinicaltrials.gov/study/NCT04671433 (accessed on 5 March 2024).
- Bainbridge, J. Gene Therapy for X-Linked Retinitis Pigmentosa (XLRP)–Retinitis Pigmentosa GTPase Regulator (RPGR) (MeiraGTx UK II Ltd.). ClinicalTrials.gov Identifier: NCT03252847. Updated 7 December 2022. Available online: https://clinicaltrials.gov/study/NCT03252847 (accessed on 5 March 2024).
- Kruczek, K.; Gonzalez-Cordero, A.; Goh, D.; Naeem, A.; Jonikas, M.; Blackford, S.J.I.; Kloc, M.; Duran, Y.; Georgiadis, A.; Sampson, R.D.; et al. Differentiation and Transplantation of Embryonic Stem Cell-Derived Cone Photoreceptors into a Mouse Model of End-Stage Retinal Degeneration. Stem Cell Rep. 2017, 8, 1659–1674. [Google Scholar] [CrossRef]
- Sun, J.; Mandai, M.; Kamao, H.; Hashiguchi, T.; Shikamura, M.; Kawamata, S.; Sugita, S.; Takahashi, M. Protective Effects of Human iPS-Derived Retinal Pigmented Epithelial Cells in Comparison with Human Mesenchymal Stromal Cells and Human Neural Stem Cells on the Degenerating Retina in rd1 mice. Stem Cells 2015, 33, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J.; Alam, N.M.; Zhao, C.; Müller, C.; Saini, J.S.; Blenkinsop, T.A.; Mazzoni, F.; Campbell, M.; Borden, S.M.; Charniga, C.J.; et al. The Developmental Stage of Adult Human Stem Cell-Derived Retinal Pigment Epithelium Cells Influences Transplant Efficacy for Vision Rescue. Stem Cell Rep. 2017, 9, 42–49. [Google Scholar] [CrossRef]
- Ortin-Martinez, A.; Tsai, E.L.; Nickerson, P.E.; Bergeret, M.; Lu, Y.; Smiley, S.; Comanita, L.; Wallace, V.A. A Reinterpretation of Cell Transplantation: GFP Transfer from Donor to Host Photoreceptors. Stem Cells 2017, 35, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Ortin-Martinez, A.; Yan, N.E.; Tsai, E.L.S.; Comanita, L.; Gurdita, A.; Tachibana, N.; Liu, Z.C.; Lu, S.; Dolati, P.; Pokrajac, N.T.; et al. Photoreceptor nanotubes mediate the in vivo exchange of intracellular material. EMBO J. 2021, 40, e107264. [Google Scholar] [CrossRef]
- Gasparini, S.J.; Tessmer, K.; Reh, M.; Wieneke, S.; Carido, M.; Völkner, M.; Borsch, O.; Swiersy, A.; Zuzic, M.; Goureau, O.; et al. Transplanted human cones incorporate into the retina and function in a murine cone degeneration model. J. Clin. Investig. 2022, 132, e154619. [Google Scholar] [CrossRef]
- Ribeiro, J.; Procyk, C.A.; West, E.L.; O’Hara-Wright, M.; Martins, M.F.; Khorasani, M.M.; Hare, A.; Basche, M.; Fernando, M.; Goh, D.; et al. Restoration of visual function in advanced disease after transplantation of purified human pluripotent stem cell-derived cone photoreceptors. Cell Rep. 2021, 35, 109022. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; McLelland, B.T.; Aramant, R.B.; Thomas, B.B.; Nistor, G.; Keirstead, H.S.; Seiler, M.J. Retina Organoid Transplants Develop Photoreceptors and Improve Visual Function in RCS Rats with RPE Dysfunction. Investig. Ophthalmol. Vis. Sci. 2020, 61, 34. [Google Scholar] [CrossRef]
- Hirami, Y.; Mandai, M.; Sugita, S.; Maeda, A.; Maeda, T.; Yamamoto, M.; Uyama, H.; Yokota, S.; Fujihara, M.; Igeta, M.; et al. Safety and stable survival of stem-cell-derived retinal organoid for 2 years in patients with retinitis pigmentosa. Cell Stem Cell 2023, 30, 1585–1596.e6. [Google Scholar] [CrossRef] [PubMed]
- jCyte, Inc. Safety and Efficacy of Intravitreal Injection of Human Retinal Progenitor Cells in Adults with Retinitis Pigmentosa. ClinicalTrials.gov Identifier: NCT03073733. Updated 28 July 2023. Available online: https://www.clinicaltrials.gov/study/NCT03073733 (accessed on 5 March 2024).
- ReNeuron Limited. Safety and Tolerability of hRPC in Retinitis Pigmentosa. ClinicalTrials.gov Identifier: NCT02464436. Updated 6 July 2023. Available online: https://www.clinicaltrials.gov/study/NCT02464436 (accessed on 5 March 2024).
- Singh, R.K.; Nasonkin, I.O. Limitations and Promise of Retinal Tissue from Human Pluripotent Stem Cells for Developing Therapies of Blindness. Front. Cell. Neurosci. 2020, 14, 179. [Google Scholar] [CrossRef]
- Sun, X.Y.; Ju, X.C.; Zhao, H.F.; You, Z.W.; Han, R.R.; Luo, Z.G. Generation of Human Blood Vessel and Vascularized Cerebral Organoids. Bio-Protocol 2023, 13, e4870. [Google Scholar] [CrossRef] [PubMed]
- Busskamp, V.; Sharma, K. Advanced vascularized human stem cell-derived retinal organoids. Investig. Ophthalmol. Vis. Sci. 2023, 64, 1894. [Google Scholar]
- Fligor, C.M.; Lavekar, S.S.; Harkin, J.; Shields, P.K.; VanderWall, K.B.; Huang, K.C.; Gomes, C.; Meyer, J.S. Extension of retinofugal projections in an assembled model of human pluripotent stem cell-derived organoids. Stem Cell Rep. 2021, 16, 2228–2241. [Google Scholar] [CrossRef]
- Cooke, J.A.; Voigt, A.P.; Collingwood, M.A.; Stone, N.E.; Whitmore, S.S.; DeLuca, A.P.; Burnight, E.R.; Anfinson, K.R.; Vakulskas, C.A.; Reutzel, A.J.; et al. Propensity of Patient-Derived iPSCs for Retinal Differentiation: Implications for Autologous Cell Replacement. Stem Cells Transl. Med. 2023, 12, 365–378. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashworth, K.E.; Weisbrod, J.; Ballios, B.G. Inherited Retinal Diseases and Retinal Organoids as Preclinical Cell Models for Inherited Retinal Disease Research. Genes 2024, 15, 705. https://doi.org/10.3390/genes15060705
Ashworth KE, Weisbrod J, Ballios BG. Inherited Retinal Diseases and Retinal Organoids as Preclinical Cell Models for Inherited Retinal Disease Research. Genes. 2024; 15(6):705. https://doi.org/10.3390/genes15060705
Chicago/Turabian StyleAshworth, Kristen E., Jessica Weisbrod, and Brian G. Ballios. 2024. "Inherited Retinal Diseases and Retinal Organoids as Preclinical Cell Models for Inherited Retinal Disease Research" Genes 15, no. 6: 705. https://doi.org/10.3390/genes15060705
APA StyleAshworth, K. E., Weisbrod, J., & Ballios, B. G. (2024). Inherited Retinal Diseases and Retinal Organoids as Preclinical Cell Models for Inherited Retinal Disease Research. Genes, 15(6), 705. https://doi.org/10.3390/genes15060705