
Gut Microbiota, Human Blood Metabolites, and Esophageal Cancer: A Mendelian Randomization Study
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Method
2.1. Study Design
2.2. Data Sources
2.2.1. Genetic Instrumental Variables for Gut Microbiome
2.2.2. Genetic Instrumental Variables for Potential Mediators
2.2.3. Genetic Instrumental Variables for Esophageal Carcinoma
2.3. Statistical Analyses
2.4. Sensitivity Analyses
3. Results
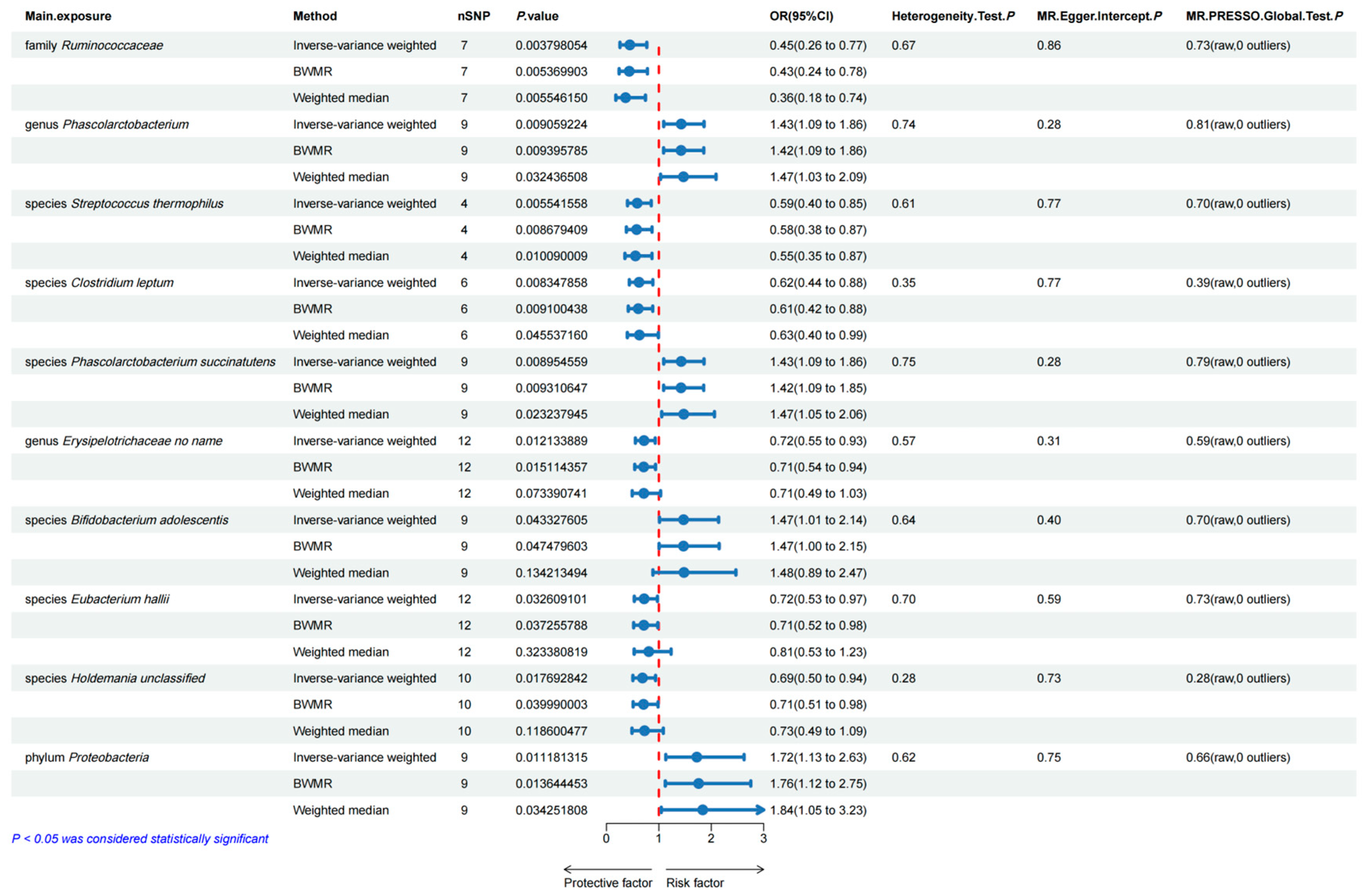
3.1. Bidirectional Two-Sample MR Analyses between Gut Microbiota and Esophageal Carcinoma
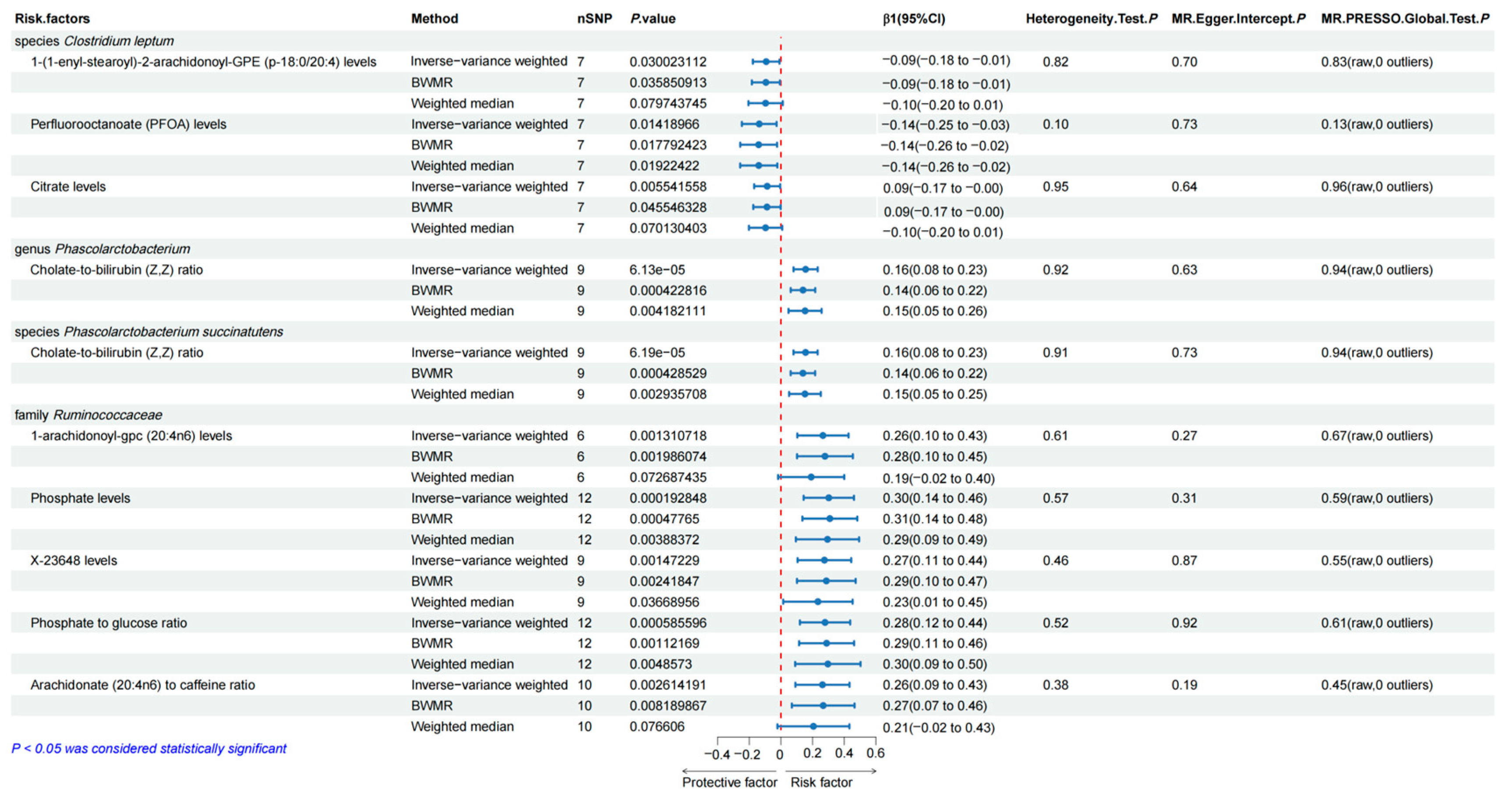
3.2. Causal Effects of the Selected Gut Microbiota on the Human Blood Metabolites
3.3. Bidirectional Two-Sample MR Analyses between Human Blood Metabolites and Esophageal Carcinoma
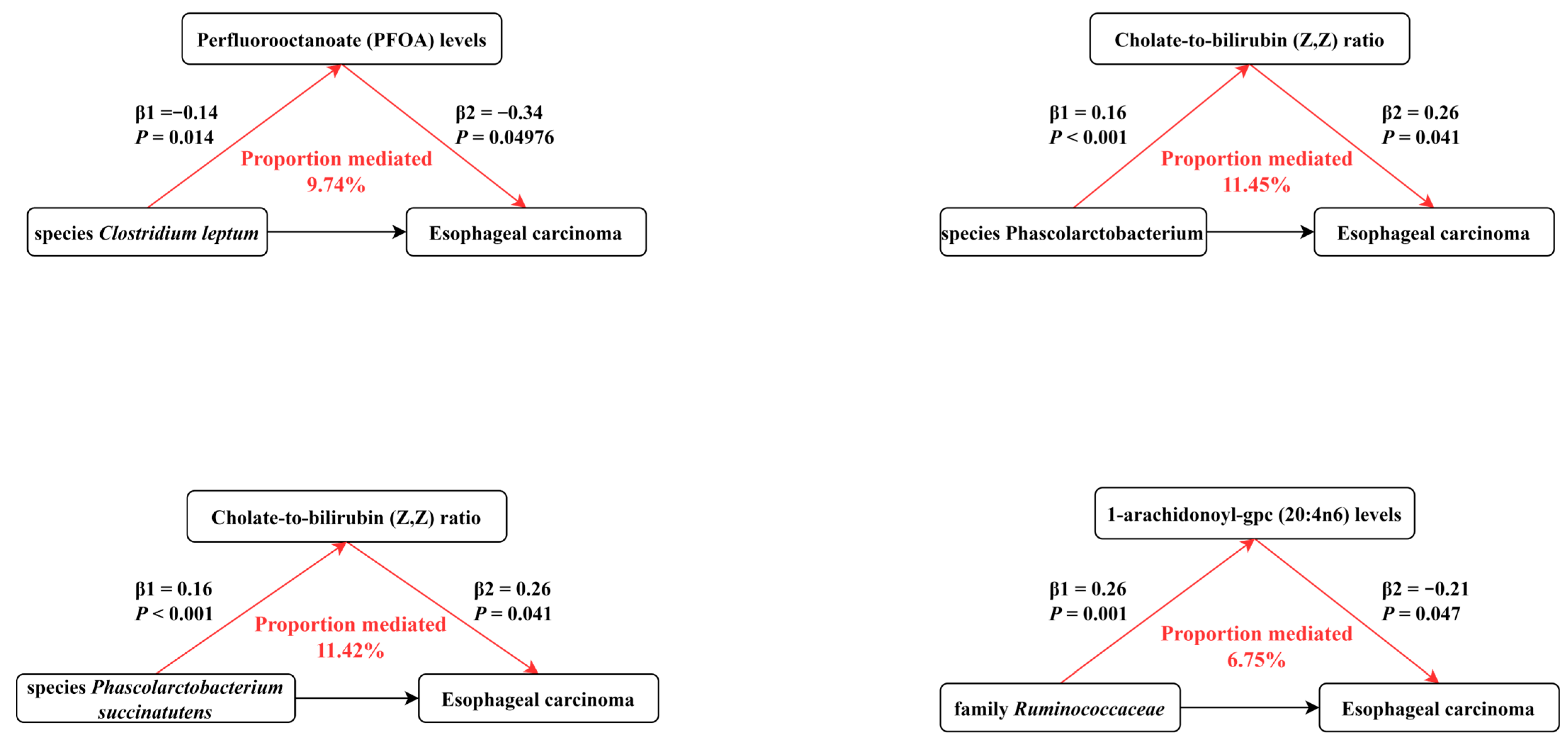
3.4. Mediation Effects of the Selected Human Blood Metabolites on Esophageal Carcinoma
3.5. Sensitivity Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Morita, F.H.; Bernardo, W.M.; Ide, E.; Rocha, R.S.; Aquino, J.C.; Minata, M.K.; Yamazaki, K.; Marques, S.B.; Sakai, P.; de Moura, E.G. Narrow band imaging versus lugol chromoendoscopy to diagnose squamous cell carcinoma of the esophagus: A systematic review and meta-analysis. BMC Cancer 2017, 17, 54. [Google Scholar] [CrossRef] [PubMed]
- NCCN Esophageal Cancer Guidelines. Available online: https://www.nccn.org/patients/guidelines/content/PDF/esophageal-patient.pdf (accessed on 5 January 2024).
- Kelly, R.J. Emerging Multimodality Approaches to Treat Localized Esophageal Cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Oppedijk, V.; van der Gaast, A.; van Lanschot, J.J.; van Hagen, P.; van Os, R.; van Rij, C.M.; van der Sangen, M.J.; Beukema, J.C.; Rütten, H.; Spruit, P.H.; et al. Patterns of recurrence after surgery alone versus preoperative chemoradiotherapy and surgery in the CROSS trials. J. Clin. Oncol. 2014, 32, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Alkasir, R.; Li, J.; Li, X.; Jin, M.; Zhu, B. Human gut microbiota: The links with dementia development. Protein Cell 2017, 8, 90–102. [Google Scholar] [CrossRef]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef]
- Muszyński, D.; Kudra, A.; Sobocki, B.K.; Folwarski, M.; Vitale, E.; Filetti, V.; Dudzic, W.; Kaźmierczak-Siedlecka, K.; Połom, K. Esophageal cancer and bacterial part of gut microbiota—A multidisciplinary point of view. Front. Cell Infect. Microbiol. 2022, 12, 1057668. [Google Scholar] [CrossRef]
- Cheung, M.K.; Yue, G.G.L.; Lauw, S.; Li, C.S.Y.; Yung, M.Y.; Ng, S.C.; Yip, H.C.; Kwan, H.S.; Chiu, P.W.Y.; Lau, C.B.S. Alterations in gut microbiota of esophageal squamous cell carcinoma patients. J. Gastroenterol. Hepatol. 2022, 37, 1919–1927. [Google Scholar] [CrossRef]
- Lin, M.Q.; Wu, Y.H.; Yang, J.; Lin, H.C.; Liu, L.Y.; Yu, Y.L.; Yao, Q.W.; Li, J.C. Gut Microbiota Characteristics Are Associated with Severity of Acute Radiation-Induced Esophagitis. Front. Microbiol. 2022, 13, 883650. [Google Scholar] [CrossRef]
- Fang, C.; Zuo, K.; Liu, Z.; Liu, Y.; Liu, L.; Wang, Y.; Yin, X.; Li, J.; Liu, X.; Chen, M.; et al. Disordered gut microbiota promotes atrial fibrillation by aggravated conduction disturbance and unbalanced linoleic acid/SIRT1 signaling. Biochem. Pharmacol. 2023, 213, 115599. [Google Scholar] [CrossRef]
- Zeng, Y.; Cao, S.; Yang, H. Roles of gut microbiome in epilepsy risk: A Mendelian randomization study. Front. Microbiol. 2023, 14, 1115014. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D. Gut microbiota: Antidiabetic drug treatment confounds gut dysbiosis associated with type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Espiridion, B.; Liang, D.; Ajani, J.A.; Liang, S.; Ye, Y.; Hildebrandt, M.A.; Gu, J.; Wu, X. Identification of Serum Markers of Esophageal Adenocarcinoma by Global and Targeted Metabolic Profiling. Clin. Gastroenterol. Hepatol. 2015, 13, 1730–1737.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, K.; Liu, G.; Wang, Y.; Xu, J.; Liu, L.; Li, M.; Shi, J.; Aa, J.; Yu, L. Metabolic Perturbation and Potential Markers in Patients with Esophageal Cancer. Gastroenterol. Res. Pract. 2017, 2017, 5469597. [Google Scholar] [CrossRef] [PubMed]
- Granja, S.; Pinheiro, C.; Reis, R.M.; Martinho, O.; Baltazar, F. Glucose Addiction in Cancer Therapy: Advances and Drawbacks. Curr. Drug Metab. 2015, 16, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Ebrahim, S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Ebrahim, S. Mendelian randomization: Prospects, potentials, and limitations. Int. J. Epidemiol. 2004, 33, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Davey Smith, G.; Ebrahim, S. What can mendelian randomisation tell us about modifiable behavioural and environmental exposures? BMJ (Clin. Res. Ed.) 2005, 330, 1076–1079. [Google Scholar] [CrossRef] [PubMed]
- Lopera-Maya, E.A.; Kurilshikov, A.; van der Graaf, A.; Hu, S.; Andreu-Sanchez, S.; Chen, L.; Vila, A.V.; Gacesa, R.; Sinha, T.; Collij, V.; et al. Effect of host genetics on the gut microbiome in 7,738 participants of the Dutch Microbiome Project. Nat. Genet. 2022, 54, 143–151. [Google Scholar] [CrossRef]
- Palmer, T.M.; Lawlor, D.A.; Harbord, R.M.; Sheehan, N.A.; Tobias, J.H.; Timpson, N.J.; Smith, G.D.; Sterne, J.A. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat. Methods Med. Res. 2012, 21, 223–242. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, T.; Pettersson-Kymmer, U.; Stewart, I.D.; Butler-Laporte, G.; Nakanishi, T.; Cerani, A.; Liang, K.Y.H.; Yoshiji, S.; Willett, J.D.S.; et al. Genomic atlas of the plasma metabolome prioritizes metabolites implicated in human diseases. Nat. Genet. 2023, 55, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Dudbridge, F.; Thompson, S.G. Combining information on multiple instrumental variables in Mendelian randomization: Comparison of allele score and summarized data methods. Stat. Med. 2016, 35, 1880–1906. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ming, J.; Hu, X.; Chen, G.; Liu, J.; Yang, C. Bayesian weighted Mendelian randomization for causal inference based on summary statistics. Bioinformatics 2020, 36, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Hemani, G.; Bowden, J.; Davey Smith, G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum. Mol. Genet. 2018, 27, R195–R208. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef]
- Mondot, S.; Kang, S.; Furet, J.P.; Aguirre de Carcer, D.; McSweeney, C.; Morrison, M.; Marteau, P.; Doré, J.; Leclerc, M. Highlighting new phylogenetic specificities of Crohn’s disease microbiota. Inflamm. Bowel Dis. 2011, 17, 185–192. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Ramirez, P.; Cadena-Ullauri, S.; Paz-Cruz, E.; Tamayo-Trujillo, R.; Ruiz-Pozo, V.A.; Zambrano, A.K. Role of the gut microbiota in hematologic cancer. Front. Microbiol. 2023, 14, 1185787. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.; Bell, R.; Klag, K.A.; Lee, S.H.; Soto, R.; Ghazaryan, A.; Buhrke, K.; Ekiz, H.A.; Ost, K.S.; Boudina, S.; et al. T cell-mediated regulation of the microbiota protects against obesity. Science 2019, 365, eaat9351. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liang, M.; Luo, J.; Peng, X. Metabolites of Clostridium leptum fermenting flaxseed polysaccharide alleviate obesity in rats. Int. J. Biol. Macromol. 2024, 264, 129907. [Google Scholar] [CrossRef]
- Gerson, L.B.; Triadafilopoulos, G. Screening for esophageal adenocarcinoma: An evidence-based approach. Am. J. Med. 2002, 113, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.S.; Chang, S.H.; Kuo, W.H.; Kuo, C.H.; Li, S.Y.; Wang, M.Y.; Chang, D.Y.; Lu, Y.S.; Huang, C.S.; Cheng, A.L.; et al. A case-control study of perfluoroalkyl substances and the risk of breast cancer in Taiwanese women. Environ. Int. 2020, 142, 105850. [Google Scholar] [CrossRef]
- Calafat, A.M.; Wong, L.Y.; Kuklenyik, Z.; Reidy, J.A.; Needham, L.L. Polyfluoroalkyl chemicals in the U.S. population: Data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000. Environ. Health Perspect. 2007, 115, 1596–1602. [Google Scholar] [CrossRef]
- Moon, J.; Mun, Y. The association between per- and polyfluoroalkyl substances (PFASs) and brain, esophageal, melanomatous skin, prostate, and lung cancer using the 2003–2018 US National Health and Nutrition Examination Survey (NHANES) datasets. Heliyon 2024, 10, e24337. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.C.; Plinsch, C.; Braeuning, A.; Buhrke, T. Activation of human nuclear receptors by perfluoroalkylated substances (PFAS). Toxicol. In Vitro 2020, 62, 104700. [Google Scholar] [CrossRef]
- Deierlein, A.L.; Rock, S.; Park, S. Persistent Endocrine-Disrupting Chemicals and Fatty Liver Disease. Curr. Environ. Health Rep. 2017, 4, 439–449. [Google Scholar] [CrossRef]
- Sen, P.; Fan, Y.; Schlezinger, J.J.; Ehrlich, S.D.; Webster, T.F.; Hyötyläinen, T.; Pedersen, O.; Orešič, M. Exposure to environmental toxicants is associated with gut microbiome dysbiosis, insulin resistance and obesity. Environ. Int. 2024, 186, 108569. [Google Scholar] [CrossRef]
- Dot, T.D.; Osawa, R.; Stackebrandt, E.J.S.; Microbiology, A. Phascolarctobacterium faecium gen. nov, spec. nov., a Novel Taxon of the Sporomusa Group of Bacteria. Syst. Appl. Microbiol. 1993, 16, 380–384. [Google Scholar] [CrossRef]
- Watanabe, Y.; Nagai, F.; Morotomi, M. Characterization of Phascolarctobacterium succinatutens sp. nov., an asaccharolytic, succinate-utilizing bacterium isolated from human feces. Appl. Environ. Microbiol. 2012, 78, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.; de Souza, R.; Kendall, C.W.; Emam, A.; Jenkins, D.J. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Al-Qadami, G.; Bowen, J.; Van Sebille, Y.; Secombe, K.; Dorraki, M.; Verjans, J.; Wardill, H.; Le, H. Baseline gut microbiota composition is associated with oral mucositis and tumour recurrence in patients with head and neck cancer: A pilot study. Support. Care Cancer 2023, 31, 98. [Google Scholar] [CrossRef] [PubMed]
- Genton, L.; Lazarevic, V.; Stojanovic, O.; Spiljar, M.; Djaafar, S.; Koessler, T.; Dutoit, V.; Gaïa, N.; Mareschal, J.; Macpherson, A.J.; et al. Metataxonomic and Metabolic Impact of Fecal Microbiota Transplantation From Patients with Pancreatic Cancer into Germ-Free Mice: A Pilot Study. Front. Cell Infect. Microbiol. 2021, 11, 752889. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, H. Compositional differences of gut microbiome in matched hormone-sensitive and castration-resistant prostate cancer. Transl. Androl. Urol. 2020, 9, 1937–1944. [Google Scholar] [CrossRef]
- Hong, J.; Behar, J.; Wands, J.; Resnick, M.; Wang, L.J.; DeLellis, R.A.; Lambeth, D.; Souza, R.F.; Spechler, S.J.; Cao, W. Role of a novel bile acid receptor TGR5 in the development of oesophageal adenocarcinoma. Gut 2010, 59, 170–180. [Google Scholar] [CrossRef]
- Morrow, D.J.; Avissar, N.E.; Toia, L.; Redmond, E.M.; Watson, T.J.; Jones, C.; Raymond, D.P.; Litle, V.; Peters, J.H. Pathogenesis of Barrett’s esophagus: Bile acids inhibit the Notch signaling pathway with induction of CDX2 gene expression in human esophageal cells. Surgery 2009, 146, 714–721; discussion 721–722. [Google Scholar] [CrossRef]
- Tamagawa, Y.; Ishimura, N.; Uno, G.; Yuki, T.; Kazumori, H.; Ishihara, S.; Amano, Y.; Kinoshita, Y. Notch signaling pathway and Cdx2 expression in the development of Barrett’s esophagus. Lab. Investig. 2012, 92, 896–909. [Google Scholar] [CrossRef]
- Zhou, Z.; Xia, Y.; Bandla, S.; Zakharov, V.; Wu, S.; Peters, J.; Godfrey, T.E.; Sun, J. Vitamin D receptor is highly expressed in precancerous lesions and esophageal adenocarcinoma with significant sex difference. Hum. Pathol. 2014, 45, 1744–1751. [Google Scholar] [CrossRef]
- Aoyama, T.; Ju, M.; Machida, D.; Komori, K.; Tamagawa, H.; Tamagawa, A.; Maezawa, Y.; Kano, K.; Hara, K.; Segami, K.; et al. Clinical Impact of Preoperative Albumin-Bilirubin Status in Esophageal Cancer Patients Who Receive Curative Treatment. In Vivo 2022, 36, 1424–1431. [Google Scholar] [CrossRef]
- Kitahama, T.; Ishii, K.; Haneda, R.; Inoue, M.; Mayanagi, S.; Tsubosa, Y. Clinical Significance of Albumin-Bilirubin Grade in Thoracic Esophageal Squamous Cell Carcinoma. J. Surg. Res. 2023, 295, 673–682. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef]
- Xu, Y.; Jing, H.; Wang, J.; Zhang, S.; Chang, Q.; Li, Z.; Wu, X.; Zhang, Z. Disordered Gut Microbiota Correlates with Altered Fecal Bile Acid Metabolism and Post-cholecystectomy Diarrhea. Front. Microbiol. 2022, 13, 800604. [Google Scholar] [CrossRef]
- De Weirdt, R.; Van de Wiele, T. Micromanagement in the gut: Microenvironmental factors govern colon mucosal biofilm structure and functionality. NPJ Biofilms Microbiomes 2015, 1, 15026. [Google Scholar] [CrossRef]
- Young, V.B.; Schmidt, T.M. Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J. Clin. Microbiol. 2004, 42, 1203–1206. [Google Scholar] [CrossRef]
- Bultman, S.J. Bacterial butyrate prevents atherosclerosis. Nat. Microbiol. 2018, 3, 1332–1333. [Google Scholar] [CrossRef]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef]
- Kurachi, M.; Kurachi, J.; Suenaga, F.; Tsukui, T.; Abe, J.; Ueha, S.; Tomura, M.; Sugihara, K.; Takamura, S.; Kakimi, K.; et al. Chemokine receptor CXCR3 facilitates CD8(+) T cell differentiation into short-lived effector cells leading to memory degeneration. J. Exp. Med. 2011, 208, 1605–1620. [Google Scholar] [CrossRef]
- Alcover, A.; Alarcón, B.; Di Bartolo, V. Cell Biology of T Cell Receptor Expression and Regulation. Annu. Rev. Immunol. 2018, 36, 103–125. [Google Scholar] [CrossRef]
- Jia, Y.; Hui, L.; Sun, L.; Guo, D.; Shi, M.; Zhang, K.; Yang, P.; Wang, Y.; Liu, F.; Shen, O.; et al. Association Between Human Blood Metabolome and the Risk of Psychiatric Disorders. Schizophr. Bull. 2023, 49, 428–443. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Xu, B.; Yang, H.; Zhu, Z. Gut Microbiota, Human Blood Metabolites, and Esophageal Cancer: A Mendelian Randomization Study. Genes 2024, 15, 729. https://doi.org/10.3390/genes15060729
Li X, Xu B, Yang H, Zhu Z. Gut Microbiota, Human Blood Metabolites, and Esophageal Cancer: A Mendelian Randomization Study. Genes. 2024; 15(6):729. https://doi.org/10.3390/genes15060729
Chicago/Turabian StyleLi, Xiuzhi, Bingchen Xu, Han Yang, and Zhihua Zhu. 2024. "Gut Microbiota, Human Blood Metabolites, and Esophageal Cancer: A Mendelian Randomization Study" Genes 15, no. 6: 729. https://doi.org/10.3390/genes15060729