Abstract
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disease that affects the motoneurons. More than 40 genes are related with ALS, and amyloidogenic proteins like SOD1 and/or TDP-43 mutants are directly involved in the onset of ALS through the formation of polymorphic fibrillogenic aggregates. However, efficacious therapeutic approaches are still lacking. Notably, heterozygous missense mutations affecting the gene coding for RNase 5, an enzyme also called angiogenin (ANG), were found to favor ALS onset. This is also true for the less-studied but angiogenic RNase 4. This review reports the substrate targets and illustrates the neuroprotective role of native ANG in the neo-vascularization of motoneurons. Then, it discusses the molecular determinants of many pathogenic ANG mutants, which almost always cause loss of function related to ALS, resulting in failures in angiogenesis and motoneuron protection. In addition, ANG mutations are sometimes combined with variants of other factors, thereby potentiating ALS effects. However, the activity of the native ANG enzyme should be finely balanced, and not excessive, to avoid possible harmful effects. Considering the interplay of these angiogenic RNases in many cellular processes, this review aims to stimulate further investigations to better elucidate the consequences of mutations in ANG and/or RNase 4 genes, in order to achieve early diagnosis and, possibly, successful therapies against ALS.
Keywords:
ALS; angiogenin; ribonucleases; RNase 4; protein aggregation; amyloidosis; neurodegeneration 1. Introduction
1.1. Neurodegenerative Diseases, Including ALS, Are Associated to Aberrant Protein Aggregation
Several devastating neurodegenerative pathologies, such as Alzheimer’s, or Parkinson’s diseases (AD and PD, respectively), as well as Amyotrophic Lateral Sclerosis (ALS) [1] share uncontrolled protein aggregation events accompanied with the formation of amyloid deposits occurring either intracellularly or, less frequently, inside the cells of the affected individuals. These deposits consist of rigid, non-ramified cross-β-sheets fibrils formed by different proteins [2,3], which can be identified by Congo Red staining [4] or Thioflavin-T (ThT)-induced fluorescence [5]. Although amyloidogenic proteins do not share primary, secondary or tertiary structures, they all assume similar supramolecular organizations [2] that differ either in composition and intima structure of the short peptides forming the cross-β-sheet structures [6], or in the structure of the fibrils formed by the whole protein [7,8]. Indeed, many protein fibrils constantly reveal high structural polymorphism, as recently reported by studies exploiting cryo-EM flanking X-ray crystallography [9,10,11,12].
In addition, more than twenty years ago, the late Prof. Dobson and his co-workers assumed that any protein, given a high concentration and destabilizing environmental conditions, can form amyloid-like fibrils [13]. Importantly, the mature amyloid fibrillar plaques can be less toxic than their precursors, i.e., the soluble, pre-fibrillar aggregates [14], and the complex role of toxic oligomers has been recently reviewed [15]. This early effect principally occurs because the oligomers can inhibit fundamental cellular processes by interacting with cell membranes, thereby inducing oxidative stress, increasing Ca2+ concentration, and leading to apoptosis [16]. However, the clinical manifestation of one particular neurodegenerative disease, such as ALS, can be paralleled by similar co-morbidity(ies), making the scenario even more complicated [17,18,19].
Indeed, ALS is not easy to be efficacioulsy counteracted because it is a multifactor disease consequent to a progressive degeneration of motoneurons taking place either in the brain or in the spinal cord. Notably, no more than about 10% of ALS cases are currently listed as familial (fALS), while up to 90% are sporadic (sALS) [20]. Nevertheless, as illustrated by van Es et al. [21], many gene-related protein mutants are involved in the disease onset [20,22,23], and mutations of the gene encoding the natively dimeric Cu/Zn superoxide dismutase-1 (SOD1) are certainly present in about 20% of fALS patients [24]. In particular, in the European population, the most frequent ALS-related mutations affect, beyond SOD1, C9orf72 intronic expansion, TARDBP, and FUS genes [25,26,27], as well as ANG [28]; however, ten years ago, more than 30 different genes were found to be associated with fALS [29].
The active role of SOD1 in ALS was discovered thirty years ago [30]. Upon undergoing aggregation [31], SOD1 affects the cytoplasm of motoneuronal cells, hence favoring the ALS income [24]. Since then, more than one hundred SOD1 mutations have been associated with ALS, with A4S, H46R, and especially G93A being the most frequently studied, but also E22G, G37R or I113T [24,30,32,33,34,35]. Non-native SOD1 aggregation, or a simple impairing trimerization, has been also associated with ALS [36,37], while SOD1 amyloidogenic oligomers displayed polymorphism in the domain formed by residues of the 28–38 segment [38]. Finally, L84F-SOD1 has very recently been associated with fALS [39], and the Cys111 residue has revealed itself to be crucial in SOD1-related ALS cases [40].
In addition, the neuronal ubiquitinated cytoplasmic inclusions of the trans-active response DNA-binding 43 kDa protein (TDP-43) [41], encoded by the TARDBP gene, are linked to ALS. Indeed, since 2006 [41], many studies have revealed the key pathogenic effect of TDP-43 in ALS [42,43,44], as well as the role of mutations in facilitating the disease onset [45,46], such as the active role of the Q/N-rich C-terminal region [47], in line with the “poly-Q expansion” protein aggregation model proposed in 1999 by Sir Max Perutz [48]. Moreover, the abnormal phosphorylation of key-Ser residues [49], as well as other structural determinants, have been investigated for their ability to promote aggregation [50,51], while TDP-43 fibrils were also recently shown to be remarkably polymorphic [10,52,53]. Nevertheless, if on one hand, polymorphism makes the challenge more difficult, on the other hand, the use of natural, or “personalized” molecules or of nanobodies contrasting the formation of harmful amyloid deposits [54,55,56,57,58,59,60], or, even better, being able to disaggregate the pre-formed fibrils [60,61,62], could hopefully become a successful therapeutic strategy. However, and notably, one single protein can also be involved in different amyloidogenic pathologies, as it is for TDP-43, whose misfolding is often found, other than ALS, together with C9orf72 intronic expansion in FrontoTemporal Dementia (FTD), which in turn drives to brain lobar degeneration [41,63].
Again, although not being the topic of this review, it must be recalled that neuroinflammation events consequent to protein aggregation are deleterious for cell life span and therefore cause the pathological manifestations of neurodegenerative diseases [64,65], like ALS [66]. Hence, the block of protein aberrant aggregation might consequently avoid, or reduce, these inflammatory consequences.
1.2. Physio-Pathological Roles of Human Angiogenin (ANG), Particularly in Neurological Contexts
In this complicated picture common to many neurodegenerative diseases, the role of the human secretory RNase 5 [67] in ALS must be highlighted beyond the proteins and factors mentioned so far. This particular basic ribonuclease is also called angiogenin [68] (ANG, Figure 1), and its connections with ALS represent the main topic of this review.
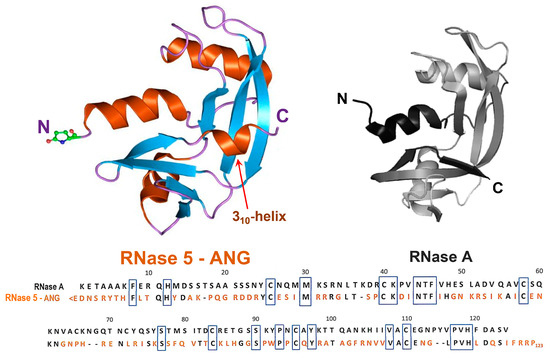
Figure 1.
Upper panel: 3D structure of angiogenin (RNase 5—ANG, pdb code 1ANG) compared with the one of RNase A (pdb code 7RSA). In both structures, the N- and C-termini are indicated. In ANG, the N-terminal pyroglutamic residue is explicitly shown. Then, α-helices are shown in orange, as well as the 310-helix tract indicated by a red arrow, while β-strands are shown in cyan and the flexible loops in purple. Lower panel: sequence alignment of the two RNase variants: where present, identity with RNase A (heading numbers refer to RNase A) is reported in black, while the different ANG residues are in orange. The black boxes indicate the residues conserved in all RNases of the super-family, comprising the H-K-H catalytic triad that in RNase A is numbered 12-41-119, while in ANG is 13-40-114.
ANG can be compared to a two-faced medal: indeed, it plays physiological roles linked to its enzymatic activity, in this way stimulating tissues’ neo-vascularization and exerting a protective action for neuronal cells, or also other cells, against stress [69]. Again, ANG lowers the proliferation of human lymphocytes, hence becoming immunosuppressive [70], but can also exert antimicrobial activity [71]. Then, it displays antiviral activity by inhibiting HIV replication [72], although it apparently favors the replication of dengue virus [73].
On the other hand, however, ANG can induce pathological effects, like the growth of tumors’ mass thanks to its angiogenic tissues “feeding” [74,75]. Again, and notably, ANG is associated to PD, and especially to ALS onset upon undergoing Loss-of-Function (LoF) missense mutations [76]. Contrarily to other proteins, like the aforementioned SOD-1 and TDP-43, the role of ANG mutants in ALS seems to be ascribable to defects occurring in its enzymatic activity or cellular localization, and not to its possible aberrant aggregation. This seems true, although it was recently detected that ANG can be induced to dimerize [77], similarly to other RNases included in the same, so-called pancreatic-type (pt-RNase) super-family [78], whose proto-type is the well-known bovine pancreatic RNase A [79].
All the effects ascribable in ALS to ANG pathogenic mutants will be deeply analyzed in the following paragraphs. Finally, mutants of human RNase 4 [80], another angiogenic enzyme included in the pt-RNase superfamily [67,78], have also been found to be involved in the onset of ALS. A short paragraph in this review will list them.
2. Human Angiogenin (ANG), a Neuroprotective Ribonuclease, Acts as a Hormone Involved in Many Physiological Pathways
2.1. Main Features of ANG and Cell-Tissue Contexts in Which Its Ribonucleolytic Activity Is Exerted
RNase 5, or human angiogenin (ANG, Figure 1 left), belongs to the mentioned secretory pt-RNase superfamily, although displaying only 33% sequence identity with RNase A (Figure 1, right) [81]. Indeed, ANG actually shares, beyond its ribonucleolytic activity [82], four fundamental features with all pt-RNases known so far. In particular, (i) a similar V-shaped, or kidney-like folding (Figure 1) [83,84], accompanied by three out of four disulfide bonds characterizing RNase A-like enzymes; (ii) a His-Lys-His catalytic triad, composed of H13-K40-H114 [83], with the two His residues located in the N- and C-termini, respectively, and Lys in the enzyme core; (iii) the ability to cleave RNA substrates preferentially on the 3′side of pyrimidines like RNase A [82], following a two-step transphosphorylation/hydrolysis mechanism [79]; (iv) a CKxxNTF consensus sequence, comprising the catalytic Lys, as well as a PVH triplet at the C-terminus comprising the second catalytic His residue [85]. However, two other features characterize ANG: (i) its N-terminal pyroglutamic acid residue, shared only with RNase 4 [80] and amphibian onconase [86], and (ii) a 310-helix tract involving the residues 117–121 that are located close to the ANG C-terminal end, which is a unique feature, absent in other pt-RNases [83].
Similarly to other secretory pt-RNases, the 5.4 kb ANG gene is located in the chromosome 14, precisely in the 14q11.2 locus [87], also encoding a leader 24AA residues-long sequence, or signal peptide preceding the mature 123 AA-residues-long ANG [88]. Hence, the numbering of ANG sequences sometimes considers these additional 24 residues as well [89]. The mRNA of ANG is present in many tissues, including the central nervous system (CNS) [87], while ANG was first characterized in 1985 as a 14.1 kDa monomer accompanied with ribonucleolytic activity [68,81] that is however lower than RNase A and other pt-RNases [82]. This reduced activity is principally ascribable to Glu116 and Phe120 [84], and, above all, to Gln117 residues that all partially obstruct the ANG active site cleft [83,90]. ANG was first characterized as a factor augmenting the tumor mass by inducing its vascularization and growth, since it was isolated and sequenced from human adenocarcinoma cell-conditioned media [68,81], or from hepatic tumors [75]. The low ribonucleolytic activity is however mandatory for ANG to exert its biological properties [91], like the activation of the growth factor in normal human tissues and plasma, or in the amniotic fluid [92,93]. Hence, it is a matter of fact to consider RNase 5 a hormone, and to call it angiogenin. Indeed, ANG promotes, at very low concentrations, and together with chemokines and factors like VEGF [94], the neo-formation of blood vessels [68,95]. It is also active in the ovarian follicle, as well as in the endometrium and placenta, with these tissues requiring extensive angiogenesis [96]. Hence, increasing levels of ANG are known to reflect the menstrual cycle phases, as well as the raised angiogenic request during pregnancy [97], and the development of the fetus blood vessels [98]. Again, ANG interacts with endothelial and smooth muscle cells, resulting in cell migration, invasion, proliferation and formation of tubular structures [99].
Last, but not least, the active role of ANG in the nervous system development and stability must be underlined: its effect starts very early, since ANG expression occurs in the axonal growth cones of mice embryos, then induces neurite growth [100]. ANG induces the activation of a Nrf2/antioxidant response in murine neurons [101], and is neuroprotective, in particular upon helping motoneuron survival [102,103].
2.2. Mechanistic Profiles and Molecular Targets of ANG Activity
From the report above, many biological activities are ascribed to ANG action [104]. These functions are exerted inside cells, in which ANG enters upon interacting with human cell-surface receptors, such as the poorly characterized 170 kDa protein factor [105], or the transmembrane heparan sulfate proteoglycan syndecan-4 that facilitates the ANG uptake into astroglia (Figure 2A) [103]. In particular, the portion comprised within the 60–68 residues forms the receptor-binding site that allows ANG to enter the cells [106]. Instead, in RNase A or RNase 1, the 60–68 peptide has a different sequence and is constrained by the Cys65-Cys72 disulfide [79]. Regardless, both RNase A and 1 can enter cells through endocytosis, but they are then sequestered by the cellular RNase Inhibitor (RI) [107], consequently forming a RI-RNase irreversible complex characterized by a KD reaching femtomolar levels [108]. A similar, very strong complex is formed with RI also by ANG [108]. However, oxidative stress destabilizes RI [109], and consequently the cytoplasmic RI-ANG complex. This unbalances the ANG subcellular distribution, thereby affecting cell growth and survival (Figure 2A) [110]. Hence, amounts of ANG can be active also in the cytosol [110], and it has been recently reported that ANG exerts a cytoplasmic protective effect on skin keratinocytes suffering oxidative stress [111,112].
Alternatively, the RI-ANG complex formation is hindered by the phosphorylation of ANG Ser28, 37 and 87 residues, or of Thr36, operated by PKC or CDK [113], hence favoring ANG nuclear translocation (Figure 2A) [113]. Once entering the nucleus thanks to the 31RRRGL35 nuclear localization signal (NLS) containing three consecutive Arg residues, ANG accumulates into the nucleolus [114], where it can cleave pRNA, also defined NoRC-associated-RNA [113]. This substrate is a non-coding RNA that regulates the rDNA transcription of ribosomal rRNA [115] through the interaction with TIP5, a factor in turn critical for chromatin remodeling [116]. The cleavage of pRNA is an uncommon transcription pathway that impairs the remodeling cascade, in turn resulting in cell growth repression [113]. Moreover, since ANG can degrade both 28S and 18S rRNAs [82], its role overpasses rRNA transcription to become a rRNA processor as well (Figure 2A,B).
Importantly, tRNAs are the main targets of ANG as well [117] (Figure 2A,B). Indeed, ANG produces non-coding tiRNA derivatives at the anticodon loop that in the nervous system can induce functional and/or dysfunctional consequences [118]. Hence, although not all tRNAs are susceptible [119], the enzymatic activity of ANG affects crucial cellular pathways, like inhibiting protein translation [120]. ANG becomes active in the cytosol under stress conditions hindering its complexation with RI [109,111,112] and permitting the enzyme to reach and cleave the anticodon loop of tRNAs (Figure 2B). In this way, 3′ and a 5′tiRNA fragments are generated, causing the inhibition of protein translation [121]. The 5′tiRNA fragment leads instead to the assembly of stress granules (SGs) [122] that in turn induce the formation of G-quadruplexes. These structures exert a neuroprotective action [123] by trapping mRNAs and provoking translation arrest (Figure 2B). All these events do in turn contrast neuronal damages [102]. In more detail, the 5′tiANG halves deriving from ANG activity interact with cytochrome C, thereby protecting cells from apoptosis during osmotic stress [124]. However, it was reported that ANG forms higher amounts of tiRNAs under oxidative rather than osmotic stress because only oxidation dramatically decreases RI activity [125]. Interestingly, it was unveiled in neuronal SH-SY5Y cells that SGs induced by ANG activity can accumulate and consequently impair the activity of the A9D mutant of progranulin, that is in turn known to exert neurodegenerative effects (Figure 2B light-red inset) [126]. Conversely, the dysregulation of the composite stress response machinery involving ANG activity can drive neuronal cells death [127]. In particular, an excessive retention of neuronal mRNA can recruit high levels of TDP-43, thereby inducing its oligomerization and fibrillogenic aggregation [128]. The same authors hypothesized that, under stress, an unbalanced mRNA translation could trigger motoneuron damages at early ALS stages.
ANG can also cleave specific sequences of the TψC-loop of tRNAs to produce short harmful tRF-3 fragments [129]. This evidence is however debated, since other authors reported that ANG likely only acts at the tRNAs anticodon loop [130], whilst other RNases, like Dicer RNase III, are certainly known to cleave the TψC-loop [131]; hence, it can be envisaged that the environmental conditions might deflect ANG from its preferential cleavage at the anticodon loop. Indeed, tRFs fragments, comprising tRF-3, are shorter than tiRNAs and behave similarly to miRNAs, being crucial in many pathological contexts, like cancer [132], or viruses [133]. Furthermore, the possibility that tRFs might impair the neuroprotective activity of the longer 5′tiRNAs could complicate the scenario involving ANG, and for all the reasons reported ANG is considered an enzyme of substrate specificity, but with divergent functional abilities [104].
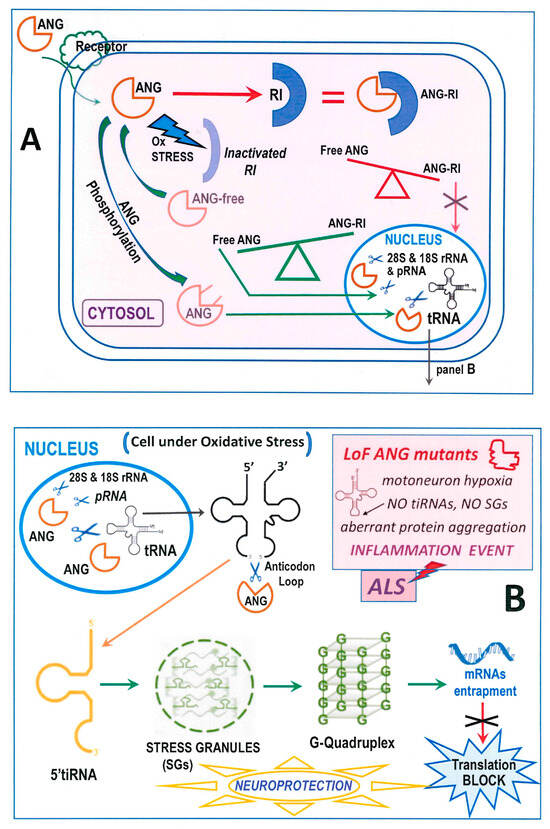
Figure 2.
Schematic view of the cellular activity of RNase 5-ANG. (A): Cell entry and destiny of ANG under normal or, alternatively, under stress conditions that can in turn favor, or not, its entering the nucleus to attack RNA substrates; “RI” = Ribonuclease Inhibitor. (B) The neuroprotective scenario ascribed to the formation of SGs and G-quadruplexes following the production of the 5′tiRNA [123], half thanks to ANG enzymatic activity. The pathological neurodegeneration events ascribable to inflammation linked to ANG LoF mutations are summarized in the light-red squared inset.
Figure 2.
Schematic view of the cellular activity of RNase 5-ANG. (A): Cell entry and destiny of ANG under normal or, alternatively, under stress conditions that can in turn favor, or not, its entering the nucleus to attack RNA substrates; “RI” = Ribonuclease Inhibitor. (B) The neuroprotective scenario ascribed to the formation of SGs and G-quadruplexes following the production of the 5′tiRNA [123], half thanks to ANG enzymatic activity. The pathological neurodegeneration events ascribable to inflammation linked to ANG LoF mutations are summarized in the light-red squared inset.
2.3. Neuroprotective Activity of ANG in the Context of ALS or Other Neurodegenerative Diseases
Native ANG can also act as a neuroprotective factor in pathologic contexts of CNS: indeed, native ANG is protective for neurons undergoing hypoxia, while its pathogenic mutants are not [134], and the wt-ANG concentration was found to be higher than its normal level in the serum of patients affected by ALS [135]. Moreover, wt-ANG promotes motoneurons’ survival either in vitro or in vivo in G93A-SOD1 ALS mice [136], while pathogenic ANG mutants do not [102]. Then, ANG undergoes selective uptake in astroglia where it can cleave RNA, contrarily to its K40I active-site mutant. This activity mediates the protection of neuronal cells in astroglia, hence highlighting the paracrine effect exerted by ANG [103]. Therefore, both ANG stability and enzymatic activity are mandatory for its protective profile, since it has been reported that poorly stable or low-activity ANG variants correlate with the early onset of ALS [137]. Surprisingly, however, the same study revealed that the loss of ANG stability and activity is proportional with a long ALS duration, while active ANG was shown to accelerate the effects of ALS and to shorten patients’ life-span [137].
The picture is therefore more complicated than hoped, and further investigations should clarify if the role of ANG in the disease can be beneficial or detrimental as a function of the pathology’s time course.
3. Impaired Levels of ANG Expression, and/or Missense Inactivating ANG Mutations Favor ALS Onset
3.1. General Aspects Depicting the Role of ANG in Neurodegenerative Pathologies
Several pathogenic mutants of many proteins are involved in the onset of ALS. The most important mutants of SOD-1 and TDP-43 have already been mentioned in a previous paragraph, with all of them undergoing extensive and uncontrolled amyloidosis directly involved in ALS-related cellular damage. In this paragraph, attention is devoted to the role of ANG in neurodegenerative pathogenesis, in particular on pathogenic mutants that, since about twenty years ago, were associated to ALS [138], as well as in some cases also with PD [76], but this falls out of the focus of this review.
We already described how native, wt-ANG can cleave the anticodon loop of tRNAs, thereby forming 5′tiRNA fragments being crucial to assemble neuroprotective SGs. However, since in humans more than 500 tRNA genes decode 61 tRNA codons, some of them can be precursors of non-coding short RNA fragments whose precise role needs further analysis [139]. Furthermore, many tRNAs undergo stabilizing post-translational modifications [140], such as an anticodon loop methylation, or ribose modifications that in turn impair the interaction with ANG. In this way, this peculiar RNase becomes unable to generate the mentioned 5′ti-RNA neuroprotective fragments [127,141]. Interestingly, an accurate review centered on the importance of small tRNA fragments in neurodegenerative disorders has been reported by Fagan et al. [118].
Pathogenesis, however, can arise from coupling a susceptible tRNA substrate with an inactive ANG variant. Indeed, although wt-ANG cleaves tRNAs under stress conditions inactivating RI [109], its main destiny is to be addressed into the nucleolus to trigger multistep pathways in turn inducing the neo-formation of vessels, either in healthy contexts or in cancer [68]. Therefore, the block of ANG activity to avoid cancer development would be likely accompanied with deleterious consequences, like the deletion of beneficial neuronal effects.
3.2. Pathogenic Mutations of ANG Related to Neurodegenerative Diseases, in Particular ALS
ANG exerts its RNase-related angiogenic activity in the nucleolus. Therefore, ANG mutations impairing its translocation into the nucleus, or that heavily limit or nullify its ribonucleolytic activity, surely hinder the neo-formation of blood vessels. Instead, if ANG were to suffer mutations impeding its interaction with RI, this might be potentially beneficial. However, as reported earlier, a higher or persistent RNase activity consequent to tRNA hypo-methylation for example [127] or to the inability of RI to block ANG also under normal conditions, could on one hand enhance the formation of SGs, but on the other hand affect the mRNA long-time retention that favors aberrant TDP-43 aggregation [128]. Consequently, the ribonucleolytic activity of ANG, and therefore its concentration and cellular localization, must be carefully balanced.
Coming back to ANG variants, nearly the totality of the pathogenic mutations induce LoF consequences. These mutations, located in the ANG gene, have often been associated with familial forms of neurodegenerative disorders, in particular ALS and PD [142,143]. The initial consideration that wt-ANG is highly concentrated in the serum of ALS patients is particularly interesting [135], but this abundance was first interpreted as a defense response exerted to protect neurons and motoneurons in an already compromised context ascribable to other factors [102,103].
Instead, mutations affecting ANG were associated to ALS [28,144]. Additional studies listed increasing numbers of ALS-related ANG mutations, either in the coding region of the signal peptide or of the mature enzyme, as visible in Figure 3 and reported in Table 1. The first mutations associated to the onset of both fALS and sALS were Q12L, K17I/E, R31K, K40I, and I46V, all affecting mature ANG: these were found by Greenway et al. in Scottish, Irish, Irish/Scottish, and Irish/English patients, while C39W-ANG was registered in a wider European context [138]. The same authors hypothesized that ANG mutations might cause autosomal dominant ALS with low penetrance, while the I46V mutation was found also in some Italian ALS patients [145].
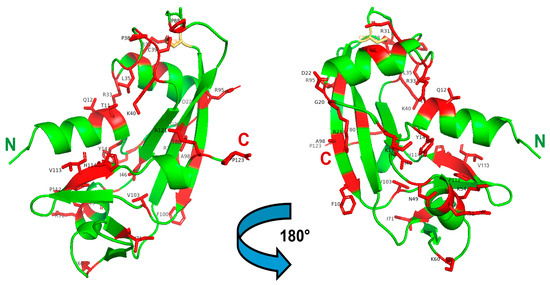
Figure 3.
3D structure of ANG highlighting the registered ALS-related mutations. Left: front-oriented visualization; Right, 180°-turn back-oriented visualization. The 30 AA residues mutated in mature ANG, and listed in Table 1, are highlighted in red and labeled in black. The C92 residue that forms a disulfide with C39, in turn affected by the C > W pathogenic mutation, is shown in yellow. The N- and C-termini of the enzyme are labeled in green and red, respectively. The Figure was built with PyMol 2.4.1.
Crabtree et al. reported that all ANG mutants, except R31K, retain low RNase activity [146], since Q12L and especially K40I directly affect the catalytic site. Instead, this effect is indirectly exerted by C39W- and I46V-ANG, that are the less thermally stable variants. The K17I/E mutants were instead hypothesized to affect peripheral catalytic subsites, like in RNase A [79,147]. Then, although the R31K variant is enzymatically active, the however conservative R-to-K mutation resulted to be detrimental because the mutation affects the Arg triplet located in the NLS 31RRRGL35 peptide. In the same year, Wu et al. reported that P(-4)S, K17I, S28N and P112L ANG mutations, the first affecting the signal peptide, were found to be LoF, i.e., not angiogenic, in North American ALS patients [148]. Additionally, P112L-ANG was only somewhat enzymatically active, but unable to enter the nucleus, while the contrary was found to be true for K17I-ANG. Instead, S28N-ANG was again unable to enter the nucleus, although it retained a partial RNase activity. It also showed propensity to dimerize, and this latter feature was considered not detrimental for RNase activity [148]. Thereafter, other ANG mutations were associated with ALS in different populations, and synonymous mutations were detected as well, like 132C->T coding for G20G [149] or 497T->G for G86G [145]. In 2008, one Italian study informed of an ALS case accompanied with the M(-24)I mutation in the ANG signal peptide [150], while Gellera et al. involved a larger cohort of Italian ALS patients. Beyond G20G and M(-24)I, other ANG mutations encoding for F(-13)S and again P(-4)S, as well as the V113I and H114R mutations, were found in mature ANG [149]. H114R directly affects one catalytic residue, obviously inducing LoF consequences, while the V113I mutation modifies a residue inserted in the 112PVH114 tract that is conserved in all pt-RNases, although the corresponding variant was not functionally tested [85]. In the same year, some French patients presented, beyond the known I46V, a R121H mutation affecting the last residue of the ANG 310-helix, hence likely altering the conformation of the enzyme catalytic cleft [151]. Again, three new, not further analyzed ALS-related ANG mutations, i.e., L35P, N49S and K60E, were discovered in Asian patients [152], while in 2009 the K54E-ANG mutant was discovered in a German patient [153]. McLaughlin et al. then analyzed how ANG levels and ANG genotypes dysregulation are crucial in Irish, Swedish and Polish ALS patients [154].
One year later, the R121C-ANG mutation, predicted to be harmful and reported as R145C because of the preceding 24AA signal peptide [88], was found in a SOD1 G93D sALS Italian patient. This was the first case of the co-presence of mutations affecting both SOD1 and ANG genes, thus inducing a speed-up of the disease progression [89]. The role of wt- or of ANG mutants in SOD1-related ALS contexts was therefore further analyzed [35,136]. Again, van Es et al. listed all the European and American nonsynonymous ANG mutations found in ALS, but also in PD patients: in particular, F(-13)L, G(-10)D, P(-4)Q, again K54E, T80S, and F100I [76], while ANG mutations coding for Y14H, D22G- and V103I-ANG were reported by two other ALS-related studies [155,156].
Then, Aparicio-Erriu and Prehn discussed the ANG paracrine activity occurring under stress conditions, like hypoxia or starvation, and on its impairment caused by ANG pathogenic mutations. They confirmed that secreted ANG accumulates in SGs (Figure 2B), thereby protecting motoneurons upon inducing an altered RNA metabolic profile in astrocytes [157]. The next year, new rare ANG mutations, i.e., G20S, P38R, R51H, P88H, R95L, A98V, and P123L emerged from other ALS patients [158]. Their possible role in affecting the conformational switch of the crucial catalytic H114 residue, as well the folding of the 31RRRGL35 nuclear localization ANG tract, was analyzed [159]. As a further consideration, it is worth mentioning that, while many cited ANG mutations are non-conservative, some of them involve Pro residues that can in turn affect proteins’ folding, as it occurs for example in RNase 1 [160].
Finally, three other rare ANG mutations, T11S, R21G, and I71V, respectively, emerged in Indian ALS patients [161,162,163], the second being LoF for either ribonucleolytic activity or nuclear translocation [162]. Again, the R33W mutation affecting the NLS 31RRRGL35 peptide was detected in the ANG gene of a Hungarian ALS patient [164].

Table 1.
Nonsynonymous RNase 5 (ANG) mutations related to the onset of ALS.
Table 1.
Nonsynonymous RNase 5 (ANG) mutations related to the onset of ALS.
| N | Residue Mutated | Country (ies) | Functional Consequences | Reference(s) |
|---|---|---|---|---|
| 1 | M(-24)I | Italy | Not Determined | Conforti FL et al., 2008 [150] |
| 2 | F(-13)L | Germany/America | Not Determined | van Es MA et al., 2011 [76] |
| 2 bis | F(-13)S | Italy | Not Determined | Gellera C et al., 2008 [149] |
| 3 | G(-10)D | Europe/America | Not Determined | van Es MA et al., 2011 [76] |
| 4 | P(-4)Q | Europe/America | Not Determined | van Es MA et al., 2011 [76] |
| 4 bis | P(-4)S | North America | Not Determined | Wu et al., 2007 [148]/Gellera et al., 2008 [149] |
| 5 | T11S | India | Negative influence to catalytic residue H114 | Padhi AK et al., 2019 [161] |
| 6 | Q12L | Scotland/Ireland | LoF *: enzymatic activity | Greenway MJ et al., 2006 [138] |
| 7 | Y14H | U.S.A. | Not reported | Brown JA et al., 2012 [155]/Zou ZY et al., 2012 [156] |
| 8 | K17I | IRE/Scotland/North America/GER/BEL/NED | LoF: enzymatic activity | Greenway et al., 2006 [138]/Wu et al., 2007 [148] |
| 8 bis | K17E | Ireland/Sweden | Partial loss enzymatic activity | Greenway MJ et al., 2006 [138] |
| 9 | G20S | Not Reported | Probable enzyme activity loss | Padhi AK et al., 2013 [158] |
| 10 | R21G | India | LoF: partial enzymatic activity + nuclear translocation | Narain P et al., 2019 [162] |
| 11 | D22G | U.S.A. | LoF: enzymatic activity | Brown JA et al., 2012 [155] |
| 12 | S28N | North America | LoF: phosphorylation hinder/ Partial enzymatic activity LOSS | Wu D et al., 2007 [148]/Fasoli S et al., 2021 [77] |
| 13 | R31K | Ireland/England | LoF: NLS impairment | Greenway, 2006 [138]/Crabtree, 2007 [146] |
| 14 | R33W | Hungary | LoF: NLS impairment | Tripolski K et al., 2019 [164] |
| 15 | L35P | Asia | LoF: partial enzymatic activity | Takahashi Y et al., 2008 [152] |
| 16 | P38R | Not Reported | Probable enzymatic activity loss | Padhi AK et al., 2013 [158] |
| 17 | C39W | Europe | LoF: enzymatic activity and folding | [138] Greenway MJ et al., 2006 |
| 18 | K40I | Ireland/England | LoF: catalytic site affected | Greenway MJ et al., 2006 [138] |
| 19 | I46V | Scotland/ITA/FRA/ GER/SWE | LoF: vdW contacts and enzymatic activity | Greenway, 2006 [138]/Corrado, 2007 [145]/ Paubel et al., 2008 [151] |
| 20 | N49S | Asia | LoF: enzymatic activity | Takahashi Y et al., 2008 [152] |
| 21 | R51H | Not reported | Not reported | Padhi AK et al., 2013 [158] |
| 22 | K54E | Germany/America | LoF: probably nuclear translocation | Fernandez-Santiago, 2009 [153]/van Es, 2011 [76] |
| 23 | K60E | Asia | No loss of enzymatic activity; not determined | Takahashi Y et al., 2008 [152] |
| 24 | I71V | India | Negative influence to catalytic residue H114 | Padhi AK et al., 2021 [163] |
| 25 | T80S | Nederland/America | LoF: vdW contacts and enzymatic activity | van Es MA et al., 2011 [76] |
| 26 | P88H | Not reported | Probable enzymatic activity loss | Padhi AK et al., 2013 [158] |
| 27 | R95L | Not reported | Probable enzymatic activity loss | Padhi AK et al., 2013 [158] |
| 28 | A98V | Not reported | Not Determined | Padhi AK et al., 2013 [158] |
| 29 | F100I | Nederland/America | Partial enzymatic activity LOSS + flexibility | van Es et al., 2011 [76]/Zou ZY et al., 2012 [156] |
| 30 | V103I | China | Partial enzymatic activity LOSS + hydrophobicity | Zou ZY et al., 2012 [156] |
| 31 | P112L | North America | Partial enzymatic activity LOSS | Wu D et al., 2007 [148] |
| 32 | V113I | Italy | Steric hindrance for substrate bind | Gellera C et al., 2008 [149] |
| 33 | H114R | Italy | LoF: catalytic site affected | Gellera C et al., 2008 [149] |
| 34 | R121H | France | Gain of Function | Paubel A et al., 2008 [151] |
| 34 bis | R121C | Italy (+G93D-SOD1) | Gain of Function | Luigetti M et al., 2011 [89] |
| 35 | P123L | Not reported | Not determined | Padhi AK et al., 2013 [158] |
* LoF: Loss of Function.
3.3. Structure-Function Investigations Related to the Neuronal Pathogenic Effects of ANG Variants
In 2012 and 2017, the group led by K.R. Acharya carried out two pivotal studies reporting detailed 3D structure-function data affecting many of the mentioned ALS-related ANG mutants [165,166]; some ANG residues were already known to be crucial catalytic subsites stabilizing the interaction(s) of the enzyme with the substrate(s) [84].
Notably, many of these key residues exhibited conformational flexibility and dual conformations that are affected by LoF pathogenic mutations, thereby justifying the decreased RNase activity of ANG, or its inability to be translocated into the nucleus, or again to induce the formation of SGs. Instead, the R121H/C variants exhibited increased RNase activity [165], as was confirmed later in our studies [77]. Interestingly, the increased RNase activity of the R121H/C-ANG mutants is likely ascribable to the decreased hindrance caused by the modified side-chain. This affects the interaction of the 121 AA-residue with Ser118 [165,166], which is in turn part of the B2 catalytic subsite [83]. The apparently paradoxical pathogenic effect due to an ANG-augmented catalytic activity was tentatively associated with the increased effect induced by R121H-ANG in neurite cultures with respect to the wt [165]. Alternatively, the already cited studies performed by Colombrita et al., or by Aluri et al. indicate that both the persistence and the increased RNase activity in ALS patients could switch the effect of ANG from protective to detrimental [128,137].
Thiyagarajan et al. also reported that the Q12L mutation affects crucial interactions with L10 and T44 [165], that are in turn part of the ANG P1 catalytic subsite involved in the phosphodiester bond cleavage [83], while the more frequent I46V mutation deletes many van der Waals contacts involving some residues of the catalytic site located in the ANG amino-terminal α-helix [165]. A similar loss of vdW contacts, in this case involving the T44 subsite, is caused by the T80S mutation too [166]. Instead, the additional methyl group of the V113I mutant sterically hinders the substrate binding, accounting for the relative decreased RNase activity. Moreover, the P112L mutation, beyond its closeness to the B2 subsite interacting with a purine base located on the opposite site of the scissile bond [83], turned out to also affect the interaction with NLS, therefore impairing ANG nuclear translocation similarly to K17I-ANG [165]. This latter aspect was also associated to the effect exerted by the S28N mutation [165], considering that the corresponding variant loses the interaction with Arg32, which is part of the NLS 31RRRGL35 sequence [114]. Instead, both F110I- and V103I-ANG displayed altered flexibility, or stabilizing interactions involving key enzyme regions, respectively [166]. Finally, the mutations of catalytic site residues, i.e., K40I, and H114R, or directly affecting the NLS sequence, like R31K, or again the stability of the enzyme, like K17I/E and C39W, were easily associated with LoF consequences [165].
Importantly, the already cited study carried out by Hoang and Raines revealed that the pathogenicity of S28N-ANG mutant can be ascribed to its scarcely phosphorylated level in the cell, thereby resulting in the cytosolic RI-ANG complexation, that in turn hinders the enzyme nuclear translocation [113]. Afterwards, the analysis of C39W and R121C ANG mutations inducing the hindering of SGs formation that in turn blocks the neuroprotective G-quadruplexes deriving from 5′-tiRNAAla fragments [123], suggested that the more active R121C-ANG may induce a partial entrapment of the fragments as a duplex form, thereby retarding the G-quadruplex formation [167]. However, additional investigations are necessary to confirm this latter hypothesis.
4. Possible Physio-Pathological Effects of ANG Self-Association
4.1. Oligomerization of Secretory pt-RNases and Their Potential Amyloidogenic Role
Some secretory basic endoribonucleases, in particular the most studied bovine pancreatic monomeric RNase A [168], can be induced to form dimers [169,170], trimers [171,172], and larger oligomers [171,172,173] through the intertwining, or the so-called 3D domain swapping (3D-DS) [174] of the N- and/or C-termini [169,170] of two or more protomers [169,170,172,173]. At least two conformers per oligomer are formed [169,170,172,173,174], all of them retaining or augmenting the ribonucleolytic activity of native RNase A [175], since their active site is reconstituted by the diverse subunits undergoing 3D-DS [169,170,172,173]. Then, the RNase A oligomers often display peculiar biological actions, in particular an anti-tumor activity exerted both in vitro and in vivo [175,176] principally because they can sterically evade RI [177].
These oligomers share some structural features [169,170] with amyloidogenic proteins: in fact, RNase A mutants displaying a Gly2–6 peptide elongation between the N-term-swapped domain and the protein core, or a Gln10 extension before the C-terminus, spontaneously produce amyloid-like fibrils through 3D-DS [178,179], similarly to proteins undergoing fibrillation through the same mechanism [180].
Although wt-RNase A is not amyloidogenic per se, some short peptides of it spontaneously form “steric-zipper cross-β-spine” polymorphic structures [6,181]. However, and notably, fibrils have also recently been detected from wt-RNase A when the enzyme overpasses the supersaturation barrier [182], or when it acts in macromolecular crowding environments [183], that can in turn also affect the extent of RNase A-controlled oligomerization [184]. Again, the RNase A N-term-swapped oligomers, whilst neither the C-term-swapped ones, nor the monomer, can promote the slow formation of “super-aggregates” [185] that resemble the protofibrils detected in crowding contexts [183].
Importantly, 3D-DS oligomerization also involves other pt-RNases. Indeed, human pancreatic RNase 1 can reach an even higher self-association extent than bovine RNase A [186], while some mutants constitutively form N-term-swapped dimers [187,188], in some cases potentially prone to undergo fibrillization [188]. These RNase 1 mutant N-swapped dimers of RNase 1 are similar, but not identical to bovine seminal (BS)-RNase, i.e., the unique natively N-swapped and antitumor dimer of the pt-RNase superfamily [189,190]. Then, the eosinophil cationic protein (ECP), i.e., RNase 3 [67], a variant that can exert neurotoxicity [191], displays a N-terminal region prone to undergo amyloidosis [192]. Again, another constitutively monomeric but antitumor RNase, i.e., the already cited amphibian onconase [86], folds similarly to RNase A despite having only 30% sequence identity [193], and is also able to form cytotoxic dimers through the swapping of its N-termini [194,195].
Importantly for the topic of this review, also ANG, or RNase 5, was recently shown to dimerize through the swapping of its N-termini [77]. In addition, a couple of ANG mutants found in patients affected by ALS or PD displayed to dimerize at a slightly higher extent than the wt [77]. Hence, a direct role of ANG pathogenic mutants in the formation of harmful neurodegenerative aggregates and fibrils could be at first envisaged. However, in the previous paragraphs, the effects of ANG in ALS onset and development were depicted to be multifaced and complicated.
4.2. Effects of ANG Oligomerization on Its Enzymatic Properties and Pathological Consequences like ALS
Wt-ANG was analyzed for its tendency to oligomerize similarly to RNase A [175] in parallel with two ALS-related ANG mutants, in particular the less enzymatically active S28N-ANG variant, that had been previously considered prone to dimerize [148], and the more enzymatically active R121C-ANG: either wt or the two ANG variants were shown to dimerize through the swapping of their N-termini, as confirmed by different experimental approaches [77], while, notably, both mutants dimerized at a higher extent than the wt. Again, all dimers maintained an enzymatic activity similar to the one of their relative monomers. Instead, neither the ANG mutants nor the wt underwent massive aggregation or fibrillation [77].
Hence, although many other mutants could and should be tested, this result suggests that ANG mutants do not produce harmful aggregates, differently from the previously mentioned SOD1 and TDP-43 pathogenic variants. However, since the conservation of the structural and functional properties of ANG can be dramatically modified by even slight modifications related to particular mutations [165], the different propensity to form dimers might somehow modify the functional properties of the enzyme in vivo, thereby affecting either ANG nuclear translocation or enzymatic efficacy. Incidentally, it has been recently reported that ANG can form a non-swapped dimer upon associating with a ds-RNA duplex, although this event has been registered only in vitro [196]. Nevertheless, the structural arrangements consequent to the interaction of ANG with RNAs, and the possible formation of dimeric or oligomeric derivatives could somehow affect the properties of the native enzyme. To this end, I recall here that a nonnative SOD1 trimer involved in ALS displayed to be toxic to motoneurons, contrarily to the SOD1 native dimer [37].
Finally, a structural model depicting the cytoplasmic interaction of ANG with the proliferating cell nuclear antigen (PCNA) [197] indicated that three ANG subunits separately interact with each one of the subunits of a cyclic PCNA trimeric assemble [198]. The authors suggest that this particular supramolecular assembly might further depict the biological roles of both ANG and PCNA in the cytoplasm. However, and finally, all these spot-like findings involving ANG oligomerization, or supramolecular organization, require further investigation, especially in the context of ALS.
5. RNase 4 Mutants Involved in ALS Onset and Development
Although this review is principally centered on the ANG-ALS connection, some other mutants of RNase 4, another member of the pt-RNase superfamily, were found to favor the ALS onset [199]. Being only 119 AA-residues long [80], RNase 4 is the smallest member of the eight “canonical” human pt-RNase groups numbered 1–8 [67]. It displays about 40% identity sequence either with both RNase A and RNase 1, or with ANG, as well as a conserved H12-K40-H116 catalytic triad [80], and it is characterized by all the features characterizing pt-RNases, like, above all, a similar V-shaped 3D structure [200].
RNase 4 is selective for 3′-uridines [200] and displays a promoter peptide, as well as a N-terminal residue a pyroglutamic acid, both features the same as ANG, with which it is also co-expressed [201].
Again, RNase 4 is protective against urinary inflammations [202]; however, conversely, it was found to favor prostate cancer cell proliferation [203].
Comparably to ANG, RNase 4 is an important angiogenic [204] and neuroprotective factor [199], Neuroprotectivity is surprisingly exerted also by its K40A mutant, considering that it retains only a slight ribonucleolytic activity [199]. Instead, the T(-13)S, R10W, E48D, V75I, and A98V mutations affecting RNase 4 have been surely associated with sALS [199]. The molecular dynamics of these mutants have been subsequently analyzed to better elucidate their structural features and their role in favoring the onset of ALS [205].
The RNase 4 gene is also affected by other LoF ALS-related mutations: in particular, D2E, N26K, T79A, and G119S [161]. Again, M29I, R31T, R32W, H72P, and R95W are LoF mutations, while the other three impair the interaction of the substrate(s) with the catalytic H116 residue [206]. No other studies further analyzed the RNase 4 role in ALS, but the ones performed often reported structure-function relationships similar to ANG-ALS.
6. Conclusions and Possible Translation to Therapeutic Approaches
The present review illustrates how the biochemical properties of ANG are at the same time peculiar and difficult to finely regulate as well. The hormonal active role of RNase 5 ANG in the neo-formation of vessels overpasses its ribonucleolytic activity, that must be in turn precisely tuned to permit the enzyme to exert its physiological activities.
Indeed, LoF mutations affecting ANG (and also RNase 4) listed here can be crucial in triggering neurodegeneration. In particular motoneuron hypoxia and death, that in turn pave the way to the ALS onset, together, or in parallel with other factors.
We additionally illustrated, however, that an excessive ANG ribonucleolytic activity can also become detrimental for patients’ survival, especially when they are affected by ALS forms that are associated with SOD1 or TDP-43 pathogenic mutants [137]. Nevertheless, studies analyzing this latter aspect are still rare and further investigations are required to better understand how the neuroprotective effects of ANG can be correctly balanced to avoid its undesired conversion to toxic, pathological consequences.
All these arguments suggest that successful strategies devoted to therapeutically counteract the harmful ANG-ALS effects are not easy to be planned. However, since ANG is certainly pathogenic upon suffering mutations, like SOD1 and TDP-43, a gene-therapy approach could be promising. Certainly, a wide genetic screening could reach individuals prone to be affected by the disease, although it would be very difficult and expensive to plan a large-scale analysis. Hence, a narrowed approach involving people affected by other ALS-inducing factors could better focus a successful targeted strategy.
However, and finally, additional studies are required to better comprehend the positive or negative contribution of ANG in ALS and to state its correct and balanced sub-cellular distribution permitting to avoid a switch from physiological to pathological effects.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Ludolph, A.C.; Brettschneider, J.; Weishaupt, J.H. Amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2012, 25, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999, 24, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Hughes, M.P.; Rodriguez, J.A.; Riek, R.; Eisenberg, D.S. The expanding amyloid family: Structure, stability, function, and pathogenesis. Cell 2021, 184, 4857–4873. [Google Scholar] [CrossRef] [PubMed]
- Klunk, W.E.; Pettegrew, J.W.; Abraham, D.J. Quantitative evaluation of congo red binding to amyloid-like proteins with a beta-pleated sheet conformation. J. Histochem. Cytochem. 1989, 37, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Naiki, H.; Higuchi, K.; Hosokawa, M.; Takeda, T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal. Biochem. 1989, 177, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Abskharon, R.; Sawaya, M.R.; Boyer, D.R.; Cao, Q.; Nguyen, B.A.; Cascio, D.; Eisenberg, D.S. Cryo-EM structure of RNA-induced tau fibrils reveals a small C-terminal core that may nucleate fibril formation. Proc. Natl. Acad. Sci. USA 2022, 119, e2119952119. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length alpha-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 2019, 26, 619–627. [Google Scholar] [CrossRef]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Abskharon, R.; Saelices, L.; Nguyen, B.A.; Lu, J.; Murray, K.A.; Kandeel, F.; Eisenberg, D.S. Cryo-EM structures of hIAPP fibrils seeded by patient-extracted fibrils reveal new polymorphs and conserved fibril cores. Nat. Struct. Mol. Biol. 2021, 28, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.A.; Singh, V.; Afrin, S.; Yakubovska, A.; Wang, L.; Ahmed, Y.; Pedretti, R.; Fernandez-Ramirez, M.D.C.; Singh, P.; Pekala, M.; et al. Structural polymorphism of amyloid fibrils in ATTR amyloidosis revealed by cryo-electron microscopy. Nat. Commun. 2024, 15, 581. [Google Scholar] [CrossRef] [PubMed]
- Fandrich, M.; Fletcher, M.A.; Dobson, C.M. Amyloid fibrils from muscle myoglobin. Nature 2001, 410, 165–166. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Rinauro, D.J.; Chiti, F.; Vendruscolo, M.; Limbocker, R. Misfolded protein oligomers: Mechanisms of formation, cytotoxic effects, and pharmacological approaches against protein misfolding diseases. Mol. Neurodegener. 2024, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef] [PubMed]
- Trojsi, F.; Siciliano, M.; Femiano, C.; Santangelo, G.; Lunetta, C.; Calvo, A.; Moglia, C.; Marinou, K.; Ticozzi, N.; Drago Ferrante, G.; et al. Comorbidity of dementia with amyotrophic lateral sclerosis (ALS): Insights from a large multicenter Italian cohort. J. Neurol. 2017, 264, 2224–2231. [Google Scholar] [CrossRef]
- Santos Garcia, D.; Suarez Castro, E.; Exposito, I.; de Deus, T.; Tunas, C.; Aneiros, A.; Lopez Fernandez, M.; Nunez Arias, D.; Bermudez Torres, M. Comorbid conditions associated with Parkinson’s disease: A longitudinal and comparative study with Alzheimer disease and control subjects. J. Neurol. Sci. 2017, 373, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Matej, R.; Tesar, A.; Rusina, R. Alzheimer’s disease and other neurodegenerative dementias in comorbidity: A clinical and neuropathological overview. Clin. Biochem. 2019, 73, 26–31. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Schymick, J.C.; Talbot, K.; Traynor, B.J. Genetics of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2007, 16, R233–R242. [Google Scholar] [CrossRef]
- Millecamps, S.; Salachas, F.; Cazeneuve, C.; Gordon, P.; Bricka, B.; Camuzat, A.; Guillot-Noel, L.; Russaouen, O.; Bruneteau, G.; Pradat, P.F.; et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: Genotype-phenotype correlations. J. Med. Genet. 2010, 47, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Chio, A.; Calvo, A.; Mazzini, L.; Cantello, R.; Mora, G.; Moglia, C.; Corrado, L.; D’Alfonso, S.; Majounie, E.; Renton, A.; et al. Parals, Extensive genetics of ALS: A population-based study in Italy. Neurology 2012, 79, 1983–1989. [Google Scholar] [CrossRef]
- Lattante, S.; Conte, A.; Zollino, M.; Luigetti, M.; Del Grande, A.; Marangi, G.; Romano, A.; Marcaccio, A.; Meleo, E.; Bisogni, G.; et al. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology 2012, 79, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Kenna, K.P.; McLaughlin, R.L.; Byrne, S.; Elamin, M.; Heverin, M.; Kenny, E.M.; Cormican, P.; Morris, D.W.; Donaghy, C.G.; Bradley, D.G.; et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J. Med. Genet. 2013, 50, 776–783. [Google Scholar] [CrossRef]
- Greenway, M.J.; Alexander, M.D.; Ennis, S.; Traynor, B.J.; Corr, B.; Frost, E.; Green, A.; Hardiman, O. A novel candidate region for ALS on chromosome 14q11.2. Neurology 2004, 63, 1936–1938. [Google Scholar] [CrossRef]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Ivanova, M.I.; Sievers, S.A.; Guenther, E.L.; Johnson, L.M.; Winkler, D.D.; Galaleldeen, A.; Sawaya, M.R.; Hart, P.J.; Eisenberg, D.S. Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Bowling, A.C.; Patterson, D.; Usdin, T.B.; Sapp, P.; Mezey, E.; McKenna-Yasek, D.; O’Regan, J.; Rahmani, Z.; Ferrante, R.J.; et al. A frequent ala 4 to val superoxide dismutase-1 mutation is associated with a rapidly progressive familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 1994, 3, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Gamez, J.; Corbera-Bellalta, M.; Nogales, G.; Raguer, N.; Garcia-Arumi, E.; Badia-Canto, M.; Llado-Carbo, E.; Alvarez-Sabin, J. Mutational analysis of the Cu/Zn superoxide dismutase gene in a Catalan ALS population: Should all sporadic ALS cases also be screened for SOD1? J. Neurol. Sci. 2006, 247, 21–28. [Google Scholar] [CrossRef]
- Sangwan, S.; Eisenberg, D.S. Perspective on SOD1 mediated toxicity in Amyotrophic Lateral Sclerosis. Postep. Biochem. 2016, 62, 362–369. [Google Scholar] [CrossRef]
- Kassa, R.M.; Bonafede, R.; Boschi, F.; Malatesta, M.; Mariotti, R. The role of mutated SOD1 gene in synaptic stripping and MHC class I expression following nerve axotomy in ALS murine model. Eur. J. Histochem. 2018, 62, 2904. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Durazo, A.; Girotto, S.; Gralla, E.B.; Martinelli, M.; Valentine, J.S.; Vieru, M.; Whitelegge, J.P. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: A possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. USA 2007, 104, 11263–11267. [Google Scholar] [CrossRef]
- Proctor, E.A.; Fee, L.; Tao, Y.; Redler, R.L.; Fay, J.M.; Zhang, Y.; Lv, Z.; Mercer, I.P.; Deshmukh, M.; Lyubchenko, Y.L.; et al. Nonnative SOD1 trimer is toxic to motor neurons in a model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, S.; Sawaya, M.R.; Murray, K.A.; Hughes, M.P.; Eisenberg, D.S. Atomic structures of corkscrew-forming segments of SOD1 reveal varied oligomer conformations. Protein Sci. 2018, 27, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Vats, A.; Ahuja, V.; Vats, K.; Khurana, S.; Vats, Y.; Gourie-Devi, M.; Wajid, S.; Ganguly, N.K.; Chakraborti, P.; et al. Functional consequences of familial ALS-associated SOD1(L84F) in neuronal and muscle cells. FASEB J. 2024, 38, e23461. [Google Scholar] [CrossRef]
- Cozzolino, M.; Amori, I.; Pesaresi, M.G.; Ferri, A.; Nencini, M.; Carri, M.T. Cysteine 111 affects aggregation and cytotoxicity of mutant Cu, Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 2008, 283, 866–874. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Soraru, G.; Orsetti, V.; Buratti, E.; Baralle, F.; Cima, V.; Volpe, M.; D’Ascenzo, C.; Palmieri, A.; Koutsikos, K.; Pegoraro, E.; et al. TDP-43 in skeletal muscle of patients affected with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. The molecular links between TDP-43 dysfunction and neurodegeneration. Adv. Genet. 2009, 66, 1–34. [Google Scholar] [PubMed]
- Lattante, S.; Sabatelli, M.; Bisogni, G.; Marangi, G.; Doronzio, P.N.; Martello, F.; Renzi, A.G.; Del Giudice, E.; Leon, A.; Cimbolli, P.; et al. Evaluating the contribution of the gene TARDBP in Italian patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2023, 30, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Corrado, L.; Ratti, A.; Gellera, C.; Buratti, E.; Castellotti, B.; Carlomagno, Y.; Ticozzi, N.; Mazzini, L.; Testa, L.; Taroni, F.; et al. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum. Mutat. 2009, 30, 688–694. [Google Scholar] [CrossRef]
- Budini, M.; Buratti, E.; Stuani, C.; Guarnaccia, C.; Romano, V.; De Conti, L.; Baralle, F.E. Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. J. Biol. Chem. 2012, 287, 7512–7525. [Google Scholar] [CrossRef]
- Perutz, M.F.; Johnson, T.; Suzuki, M.; Finch, J.T. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 1994, 91, 5355–5358. [Google Scholar] [CrossRef]
- Inukai, Y.; Nonaka, T.; Arai, T.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.E.; Akiyama, H.; Hisanaga, S.; et al. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 2008, 582, 2899–2904. [Google Scholar] [CrossRef]
- Mompean, M.; Hervas, R.; Xu, Y.; Tran, T.H.; Guarnaccia, C.; Buratti, E.; Baralle, F.; Tong, L.; Carrion-Vazquez, M.; McDermott, A.E.; et al. Structural Evidence of Amyloid Fibril Formation in the Putative Aggregation Domain of TDP-43. J. Phys. Chem. Lett. 2015, 6, 2608–2615. [Google Scholar] [CrossRef]
- Mompean, M.; Baralle, M.; Buratti, E.; Laurents, D.V. An Amyloid-Like Pathological Conformation of TDP-43 Is Stabilized by Hypercooperative Hydrogen Bonds. Front. Mol. Neurosci. 2016, 9, 125. [Google Scholar] [CrossRef]
- Guenther, E.L.; Ge, P.; Trinh, H.; Sawaya, M.R.; Cascio, D.; Boyer, D.R.; Gonen, T.; Zhou, Z.H.; Eisenberg, D.S. Atomic-level evidence for packing and positional amyloid polymorphism by segment from TDP-43 RRM2. Nat. Struct. Mol. Biol. 2018, 25, 311–319. [Google Scholar] [CrossRef]
- Carrasco, J.; Anton, R.; Valbuena, A.; Pantoja-Uceda, D.; Mukhi, M.; Hervas, R.; Laurents, D.V.; Gasset, M.; Oroz, J. Metamorphism in TDP-43 prion-like domain determines chaperone recognition. Nat. Commun. 2023, 14, 466. [Google Scholar] [CrossRef] [PubMed]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Sievers, S.A.; Karanicolas, J.; Chang, H.W.; Zhao, A.; Jiang, L.; Zirafi, O.; Stevens, J.T.; Munch, J.; Baker, D.; Eisenberg, D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 2011, 475, 96–100. [Google Scholar] [CrossRef]
- Palazzi, L.; Leri, M.; Cesaro, S.; Stefani, M.; Bucciantini, M.; Polverino de Laureto, P. Insight into the molecular mechanism underlying the inhibition of alpha-synuclein aggregation by hydroxytyrosol. Biochem. Pharmacol. 2020, 173, 113722. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, S.; Sahay, S.; Murray, K.A.; Morgan, S.; Guenther, E.L.; Jiang, L.; Williams, C.K.; Vinters, H.V.; Goedert, M.; Eisenberg, D.S. Inhibition of synucleinopathic seeding by rationally designed inhibitors. eLife 2020, 9, e46775. [Google Scholar] [CrossRef]
- Murray, K.A.; Hu, C.J.; Griner, S.L.; Pan, H.; Bowler, J.T.; Abskharon, R.; Rosenberg, G.M.; Cheng, X.; Seidler, P.M.; Eisenberg, D.S. De novo designed protein inhibitors of amyloid aggregation and seeding. Proc. Natl. Acad. Sci. USA 2022, 119, e2206240119. [Google Scholar] [CrossRef] [PubMed]
- Abskharon, R.; Pan, H.; Sawaya, M.R.; Seidler, P.M.; Olivares, E.J.; Chen, Y.; Murray, K.A.; Zhang, J.; Lantz, C.; Bentzel, M.; et al. Structure-based design of nanobodies that inhibit seeding of Alzheimer’s patient-extracted tau fibrils. Proc. Natl. Acad. Sci. USA 2023, 120, e2300258120. [Google Scholar] [CrossRef]
- Garcia-Pardo, J.; Ventura, S. Chemical targeting of amyloids. Nat. Chem. Biol. 2023, 19, 1176–1177. [Google Scholar] [CrossRef]
- Seidler, P.M.; Murray, K.A.; Boyer, D.R.; Ge, P.; Sawaya, M.R.; Hu, C.J.; Cheng, X.; Abskharon, R.; Pan, H.; DeTure, M.A.; et al. Structure-based discovery of small molecules that disaggregate Alzheimer’s disease tissue derived tau fibrils in vitro. Nat. Commun. 2022, 13, 5451. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.A.; Hu, C.J.; Pan, H.; Lu, J.; Abskharon, R.; Bowler, J.T.; Rosenberg, G.M.; Williams, C.K.; Elezi, G.; Balbirnie, M.; et al. Small molecules disaggregate alpha-synuclein and prevent seeding from patient brain-derived fibrils. Proc. Natl. Acad. Sci. USA 2023, 120, e2217835120. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Tondo, G.; Corrado, L.; Menegon, F.; Aprile, D.; Anselmi, M.; D’Alfonso, S.; Comi, C.; Mazzini, L. Neuroinflammatory Pathways in the ALS-FTD Continuum: A Focus on Genetic Variants. Genes 2023, 14, 1658. [Google Scholar] [CrossRef] [PubMed]
- Schain, M.; Kreisl, W.C. Neuroinflammation in Neurodegenerative Disorders-a Review. Curr. Neurol. Neurosci. Rep. 2017, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Xu, Y.; Liu, M.; Cui, L. The Inflammatory Puzzle: Piecing together the Links between Neuroinflammation and Amyotrophic Lateral Sclerosis. Aging Dis. 2024, 15, 96–114. [Google Scholar] [CrossRef]
- Sorrentino, S. The eight human “canonical” ribonucleases: Molecular diversity, catalytic properties, and special biological actions of the enzyme proteins. FEBS Lett. 2010, 584, 2194–2200. [Google Scholar] [CrossRef]
- Fett, J.W.; Strydom, D.J.; Lobb, R.R.; Alderman, E.M.; Bethune, J.L.; Riordan, J.F.; Vallee, B.L. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry 1985, 24, 5480–5486. [Google Scholar] [CrossRef]
- Garnett, E.R.; Raines, R.T. Emerging biological functions of ribonuclease 1 and angiogenin. Crit. Rev. Biochem. Mol. Biol. 2022, 57, 244–260. [Google Scholar] [CrossRef]
- Matousek, J.; Soucek, J.; Riha, J.; Zankel, T.R.; Benner, S.A. Immunosuppressive activity of angiogenin in comparison with bovine seminal ribonuclease and pancreatic ribonuclease. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1995, 112, 235–241. [Google Scholar] [CrossRef]
- Hooper, L.V.; Stappenbeck, T.S.; Hong, C.V.; Gordon, J.I. Angiogenins: A new class of microbicidal proteins involved in innate immunity. Nat. Immunol. 2003, 4, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Cocchi, F.; DeVico, A.L.; Lu, W.; Popovic, M.; Latinovic, O.; Sajadi, M.M.; Redfield, R.R.; Lafferty, M.K.; Galli, M.; Garzino-Demo, A.; et al. Soluble factors from T cells inhibiting X4 strains of HIV are a mixture of beta chemokines and RNases. Proc. Natl. Acad. Sci. USA 2012, 109, 5411–5416. [Google Scholar] [CrossRef] [PubMed]
- Madhry, D.; Malvankar, S.; Phadnis, S.; Srivastava, R.K.; Bhattacharyya, S.; Verma, B. Synergistic correlation between host angiogenin and dengue virus replication. RNA Biol. 2023, 20, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Kao, R.Y.; Jenkins, J.L.; Olson, K.A.; Key, M.E.; Fett, J.W.; Shapiro, R. A small-molecule inhibitor of the ribonucleolytic activity of human angiogenin that possesses antitumor activity. Proc. Natl. Acad. Sci. USA 2002, 99, 10066–10071. [Google Scholar] [CrossRef] [PubMed]
- Barcena, C.; Stefanovic, M.; Tutusaus, A.; Martinez-Nieto, G.A.; Martinez, L.; Garcia-Ruiz, C.; de Mingo, A.; Caballeria, J.; Fernandez-Checa, J.C.; Mari, M.; et al. Angiogenin secretion from hepatoma cells activates hepatic stellate cells to amplify a self-sustained cycle promoting liver cancer. Sci. Rep. 2015, 5, 7916. [Google Scholar] [CrossRef] [PubMed]
- van Es, M.A.; Schelhaas, H.J.; van Vught, P.W.; Ticozzi, N.; Andersen, P.M.; Groen, E.J.; Schulte, C.; Blauw, H.M.; Koppers, M.; Diekstra, F.P.; et al. Angiogenin variants in Parkinson disease and amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 964–973. [Google Scholar] [CrossRef]
- Fasoli, S.; Bettin, I.; Montioli, R.; Fagagnini, A.; Peterle, D.; Laurents, D.V.; Gotte, G. Dimerization of Human Angiogenin and of Variants Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 10068. [Google Scholar] [CrossRef]
- Sorrentino, S.; Libonati, M. Human pancreatic-type and nonpancreatic-type ribonucleases: A direct side-by-side comparison of their catalytic properties. Arch. Biochem. Biophys. 1994, 312, 340–348. [Google Scholar] [CrossRef]
- Raines, R.T. Ribonuclease A. Chem. Rev. 1998, 98, 1045–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.M.; Strydom, D.J. The amino acid sequence of human ribonuclease 4, a highly conserved ribonuclease that cleaves specifically on the 3′ side of uridine. Eur. J. Biochem. 1993, 217, 401–410. [Google Scholar] [CrossRef]
- Strydom, D.J.; Fett, J.W.; Lobb, R.R.; Alderman, E.M.; Bethune, J.L.; Riordan, J.F.; Vallee, B.L. Amino acid sequence of human tumor derived angiogenin. Biochemistry 1985, 24, 5486–5494. [Google Scholar] [CrossRef]
- Shapiro, R.; Riordan, J.F.; Vallee, B.L. Characteristic ribonucleolytic activity of human angiogenin. Biochemistry 1986, 25, 3527–3532. [Google Scholar] [CrossRef] [PubMed]
- Acharya, K.R.; Shapiro, R.; Allen, S.C.; Riordan, J.F.; Vallee, B.L. Crystal structure of human angiogenin reveals the structural basis for its functional divergence from ribonuclease. Proc. Natl. Acad. Sci. USA 1994, 91, 2915–2919. [Google Scholar] [CrossRef]
- Leonidas, D.D.; Shapiro, R.; Allen, S.C.; Subbarao, G.V.; Veluraja, K.; Acharya, K.R. Refined crystal structures of native human angiogenin and two active site variants: Implications for the unique functional properties of an enzyme involved in neovascularisation during tumour growth. J. Mol. Biol. 1999, 285, 1209–1233. [Google Scholar] [CrossRef]
- Smith, B.D.; Raines, R.T. Genetic selection for critical residues in ribonucleases. J. Mol. Biol. 2006, 362, 459–478. [Google Scholar] [CrossRef] [PubMed]
- Ardelt, W.; Mikulski, S.M.; Shogen, K. Amino acid sequence of an anti-tumor protein from Rana pipiens oocytes and early embryos. Homology to pancreatic ribonucleases. J. Biol. Chem. 1991, 266, 245–251. [Google Scholar] [CrossRef]
- Weiner, H.L.; Weiner, L.H.; Swain, J.L. Tissue distribution and developmental expression of the messenger RNA encoding angiogenin. Science 1987, 237, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, K.; Davie, E.W.; Strydom, D.J.; Riordan, J.F.; Vallee, B.L. Sequence of the cDNA and gene for angiogenin, a human angiogenesis factor. Biochemistry 1985, 24, 5494–5499. [Google Scholar] [CrossRef]
- Luigetti, M.; Lattante, S.; Zollino, M.; Conte, A.; Marangi, G.; Del Grande, A.; Sabatelli, M. SOD1 G93D sporadic amyotrophic lateral sclerosis (SALS) patient with rapid progression and concomitant novel ANG variant. Neurobiol. Aging 2011, 32, 1924.e15–1924.e18. [Google Scholar] [CrossRef]
- Russo, N.; Shapiro, R.; Acharya, K.R.; Riordan, J.F.; Vallee, B.L. Role of glutamine-117 in the ribonucleolytic activity of human angiogenin. Proc. Natl. Acad. Sci. USA 1994, 91, 2920–2924. [Google Scholar] [CrossRef]
- Shapiro, R.; Vallee, B.L. Site-directed mutagenesis of histidine-13 and histidine-114 of human angiogenin. Alanine derivatives inhibit angiogenin-induced angiogenesis. Biochemistry 1989, 28, 7401–7408. [Google Scholar] [CrossRef]
- Shapiro, R.; Strydom, D.J.; Olson, K.A.; Vallee, B.L. Isolation of angiogenin from normal human plasma. Biochemistry 1987, 26, 5141–5146. [Google Scholar] [CrossRef] [PubMed]
- Spong, C.Y.; Ghidini, A.; Sherer, D.M.; Pezzullo, J.C.; Ossandon, M.; Eglinton, G.S. Angiogenin: A marker for preterm delivery in midtrimester amniotic fluid. Am. J. Obstet. Gynecol. 1997, 176, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Carmeliet, P. VEGF at the neurovascular interface: Therapeutic implications for motor neuron disease. Biochim. Biophys. Acta 2006, 1762, 1109–1121. [Google Scholar] [CrossRef]
- Strydom, D.J. The angiogenins. Cell Mol. Life Sci. 1998, 54, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.P.; Grazul-Bilska, A.T.; Redmer, D.A. Angiogenesis in the female reproductive organs: Pathological implications. Int. J. Exp. Pathol. 2002, 83, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Rajashekhar, G.; Loganath, A.; Roy, A.C.; Wong, Y.C. Expression and localization of angiogenin in placenta: Enhanced levels at term over first trimester villi. Mol. Reprod. Dev. 2002, 62, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, N.; Frendo, J.L.; Guibourdenche, J.; Degrelle, S.A.; Evain-Brion, D.; Badet, J. Angiogenin expression during early human placental development; association with blood vessel formation. Biomed. Res. Int. 2014, 2014, 781632. [Google Scholar] [CrossRef]
- Gao, X.; Xu, Z. Mechanisms of action of angiogenin. Acta Biochim. Biophys. Sin. 2008, 40, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Feng, Y. A new role for angiogenin in neurite growth and pathfinding: Implications for amyotrophic lateral sclerosis. Hum. Mol. Genet. 2007, 16, 1445–1453. [Google Scholar] [CrossRef]
- Hoang, T.T.; Johnson, D.A.; Raines, R.T.; Johnson, J.A. Angiogenin activates the astrocytic Nrf2/antioxidant-response element pathway and thereby protects murine neurons from oxidative stress. J. Biol. Chem. 2019, 294, 15095–15103. [Google Scholar] [CrossRef] [PubMed]
- Kieran, D.; Sebastia, J.; Greenway, M.J.; King, M.A.; Connaughton, D.; Concannon, C.G.; Fenner, B.; Hardiman, O.; Prehn, J.H. Control of motoneuron survival by angiogenin. J. Neurosci. 2008, 28, 14056–14061. [Google Scholar] [CrossRef] [PubMed]
- Skorupa, A.; King, M.A.; Aparicio, I.M.; Dussmann, H.; Coughlan, K.; Breen, B.; Kieran, D.; Concannon, C.G.; Marin, P.; Prehn, J.H. Motoneurons secrete angiogenin to induce RNA cleavage in astroglia. J. Neurosci. 2012, 32, 5024–5038. [Google Scholar] [CrossRef] [PubMed]
- Tello-Montoliu, A.; Patel, J.V.; Lip, G.Y. Angiogenin: A review of the pathophysiology and potential clinical applications. J. Thromb. Haemost. 2006, 4, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.F.; Riordan, J.F.; Vallee, B.L. A putative angiogenin receptor in angiogenin-responsive human endothelial cells. Proc. Natl. Acad. Sci. USA 1997, 94, 2204–2209. [Google Scholar] [CrossRef] [PubMed]
- Hallahan, T.W.; Shapiro, R.; Vallee, B.L. Dual site model for the organogenic activity of angiogenin. Proc. Natl. Acad. Sci. USA 1991, 88, 2222–2226. [Google Scholar] [CrossRef] [PubMed]
- Kobe, B.; Deisenhofer, J. Mechanism of ribonuclease inhibition by ribonuclease inhibitor protein based on the crystal structure of its complex with ribonuclease A. J. Mol. Biol. 1996, 264, 1028–1043. [Google Scholar] [CrossRef] [PubMed]
- Rutkoski, T.J.; Raines, R.T. Evasion of ribonuclease inhibitor as a determinant of ribonuclease cytotoxicity. Curr. Pharm. Biotechnol. 2008, 9, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, M.; Fominaya, J.M.; Hofsteenge, J. Oxidation of sulfhydryl groups of ribonuclease inhibitor in epithelial cells is sufficient for its intracellular degradation. J. Biol. Chem. 1996, 271, 18638–18642. [Google Scholar] [CrossRef]
- Pizzo, E.; Sarcinelli, C.; Sheng, J.; Fusco, S.; Formiggini, F.; Netti, P.; Yu, W.; D’Alessio, G.; Hu, G.F. Ribonuclease/angiogenin inhibitor 1 regulates stress-induced subcellular localization of angiogenin to control growth and survival. J. Cell Sci. 2013, 126 Pt 18, 4308–4319. [Google Scholar] [CrossRef]
- Culurciello, R.; Bosso, A.; Troisi, R.; Barrella, V.; Di Nardo, I.; Borriello, M.; Gaglione, R.; Pistorio, V.; Aceto, S.; Cafaro, V.; et al. Protective Effects of Recombinant Human Angiogenin in Keratinocytes: New Insights on Oxidative Stress Response Mediated by RNases. Int. J. Mol. Sci. 2022, 23, 8781. [Google Scholar] [CrossRef]
- Culurciello, R.; Di Nardo, I.; Bosso, A.; Tortora, F.; Troisi, R.; Sica, F.; Arciello, A.; Notomista, E.; Pizzo, E. Tailoring the stress response of human skin cells by substantially limiting the nuclear localization of angiogenin. Heliyon 2024, 10, e24556. [Google Scholar] [CrossRef]
- Hoang, T.T.; Raines, R.T. Molecular basis for the autonomous promotion of cell proliferation by angiogenin. Nucleic Acids Res. 2017, 45, 818–831. [Google Scholar] [CrossRef]
- Moroianu, J.; Riordan, J.F. Identification of the nucleolar targeting signal of human angiogenin. Biochem. Biophys. Res. Commun. 1994, 203, 1765–1772. [Google Scholar] [CrossRef]
- Mayer, C.; Schmitz, K.M.; Li, J.; Grummt, I.; Santoro, R. Intergenic transcripts regulate the epigenetic state of rRNA genes. Mol. Cell 2006, 22, 351–361. [Google Scholar] [CrossRef]
- Mayer, C.; Neubert, M.; Grummt, I. The structure of NoRC-associated RNA is crucial for targeting the chromatin remodelling complex NoRC to the nucleolus. EMBO Rep. 2008, 9, 774–780. [Google Scholar] [CrossRef]
- Lee, F.S.; Vallee, B.L. Characterization of ribonucleolytic activity of angiogenin towards tRNA. Biochem. Biophys. Res. Commun. 1989, 161, 121–126. [Google Scholar] [CrossRef]
- Fagan, S.G.; Helm, M.; Prehn, J.H.M. tRNA-derived fragments: A new class of non-coding RNA with key roles in nervous system function and dysfunction. Prog. Neurobiol. 2021, 205, 102118. [Google Scholar] [CrossRef]
- Kazimierczyk, M.; Wojnicka, M.; Biala, E.; Zydowicz-Machtel, P.; Imiolczyk, B.; Ostrowski, T.; Kurzynska-Kokorniak, A.; Wrzesinski, J. Characteristics of Transfer RNA-Derived Fragments Expressed during Human Renal Cell Development: The Role of Dicer in tRF Biogenesis. Int. J. Mol. Sci. 2022, 23, 3644. [Google Scholar] [CrossRef]
- Ivanov, P.; Emara, M.M.; Villen, J.; Gygi, S.P.; Anderson, P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell 2011, 43, 613–623. [Google Scholar] [CrossRef]
- Yamasaki, S.; Ivanov, P.; Hu, G.F.; Anderson, P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell Biol. 2009, 185, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Emara, M.M.; Ivanov, P.; Hickman, T.; Dawra, N.; Tisdale, S.; Kedersha, N.; Hu, G.F.; Anderson, P. Angiogenin-induced tRNA-derived stress-induced RNAs promote stress-induced stress granule assembly. J. Biol. Chem. 2010, 285, 10959–10968. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.; O’Day, E.; Emara, M.M.; Wagner, G.; Lieberman, J.; Anderson, P. G-quadruplex structures contribute to the neuroprotective effects of angiogenin-induced tRNA fragments. Proc. Natl. Acad. Sci. USA 2014, 111, 18201–18206. [Google Scholar] [CrossRef] [PubMed]
- Saikia, M.; Krokowski, D.; Guan, B.J.; Ivanov, P.; Parisien, M.; Hu, G.F.; Anderson, P.; Pan, T.; Hatzoglou, M. Genome-wide identification and quantitative analysis of cleaved tRNA fragments induced by cellular stress. J. Biol. Chem. 2012, 287, 42708–42725. [Google Scholar] [CrossRef] [PubMed]
- Saikia, M.; Jobava, R.; Parisien, M.; Putnam, A.; Krokowski, D.; Gao, X.H.; Guan, B.J.; Yuan, Y.; Jankowsky, E.; Feng, Z.; et al. Angiogenin-cleaved tRNA halves interact with cytochrome c, protecting cells from apoptosis during osmotic stress. Mol. Cell Biol. 2014, 34, 2450–2463. [Google Scholar] [CrossRef]
- Li, S.; Chen, Y.; Sun, D.; Bai, R.; Gao, X.; Yang, Y.; Sheng, J.; Xu, Z. Angiogenin Prevents Progranulin A9D Mutation-Induced Neuronal-Like Cell Apoptosis Through Cleaving tRNAs into tiRNAs. Mol. Neurobiol. 2018, 55, 1338–1351. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Dietmann, S.; Flores, J.V.; Hussain, S.; Kutter, C.; Humphreys, P.; Lukk, M.; Lombard, P.; Treps, L.; Popis, M.; et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014, 33, 2020–2039. [Google Scholar] [CrossRef]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ender, C.; Meister, G.; Moore, P.S.; Chang, Y.; John, B. Extensive terminal and asymmetric processing of small RNAs from rRNAs, snoRNAs, snRNAs, and tRNAs. Nucleic Acids Res. 2012, 40, 6787–6799. [Google Scholar] [CrossRef]
- Su, Z.; Kuscu, C.; Malik, A.; Shibata, E.; Dutta, A. Angiogenin generates specific stress-induced tRNA halves and is not involved in tRF-3-mediated gene silencing. J. Biol. Chem. 2019, 294, 16930–16941. [Google Scholar] [CrossRef]
- Haussecker, D.; Huang, Y.; Lau, A.; Parameswaran, P.; Fire, A.Z.; Kay, M.A. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA 2010, 16, 673–695. [Google Scholar] [CrossRef] [PubMed]
- Maute, R.L.; Schneider, C.; Sumazin, P.; Holmes, A.; Califano, A.; Basso, K.; Dalla-Favera, R. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Schorn, A.J.; Gutbrod, M.J.; LeBlanc, C.; Martienssen, R. LTR-Retrotransposon Control by tRNA-Derived Small RNAs. Cell 2017, 170, 61–71.e11. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Crabtree, B.; Acharya, K.R. Human angiogenin is a neuroprotective factor and amyotrophic lateral sclerosis associated angiogenin variants affect neurite extension/pathfinding and survival of motor neurons. Hum. Mol. Genet. 2008, 17, 130–149. [Google Scholar] [CrossRef] [PubMed]
- Cronin, S.; Greenway, M.J.; Ennis, S.; Kieran, D.; Green, A.; Prehn, J.H.; Hardiman, O. Elevated serum angiogenin levels in ALS. Neurology 2006, 67, 1833–1836. [Google Scholar] [CrossRef] [PubMed]
- Crivello, M.; O’Riordan, S.L.; Woods, I.; Cannon, S.; Halang, L.; Coughlan, K.S.; Hogg, M.C.; Lewandowski, S.A.; Prehn, J.H.M. Pleiotropic activity of systemically delivered angiogenin in the SOD1(G93A) mouse model. Neuropharmacology 2018, 133, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Aluri, K.C.; Salisbury, J.P.; Prehn, J.H.M.; Agar, J.N. Loss of angiogenin function is related to earlier ALS onset and a paradoxical increase in ALS duration. Sci. Rep. 2020, 10, 3715. [Google Scholar] [CrossRef] [PubMed]
- Greenway, M.J.; Andersen, P.M.; Russ, C.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef]
- Torres, A.G.; Reina, O.; Stephan-Otto Attolini, C.; Ribas de Pouplana, L. Differential expression of human tRNA genes drives the abundance of tRNA-derived fragments. Proc. Natl. Acad. Sci. USA 2019, 116, 8451–8456. [Google Scholar] [CrossRef]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crecy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic. Acids. Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef]
- Oberbauer, V.; Schaefer, M.R. tRNA-Derived Small RNAs: Biogenesis, Modification, Function and Potential Impact on Human Disease Development. Genes 2018, 9, 607. [Google Scholar] [CrossRef] [PubMed]
- Rayaprolu, S.; Soto-Ortolaza, A.; Rademakers, R.; Uitti, R.J.; Wszolek, Z.K.; Ross, O.A. Angiogenin variation and Parkinson disease. Ann. Neurol. 2012, 71, 725–727, author reply 727-8. [Google Scholar] [CrossRef] [PubMed]
- Prehn, J.H.M.; Jirstrom, E. Angiogenin and tRNA fragments in Parkinson’s disease and neurodegeneration. Acta Pharmacol. Sin. 2020, 41, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Lafuste, P.; Carmeliet, P.; Conway, E.M. Another angiogenic gene linked to amyotrophic lateral sclerosis. Trends Mol. Med. 2006, 12, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Corrado, L.; Battistini, S.; Penco, S.; Bergamaschi, L.; Testa, L.; Ricci, C.; Giannini, F.; Greco, G.; Patrosso, M.C.; Pileggi, S.; et al. Variations in the coding and regulatory sequences of the angiogenin (ANG) gene are not associated to ALS (amyotrophic lateral sclerosis) in the Italian population. J. Neurol. Sci. 2007, 258, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, B.; Thiyagarajan, N.; Prior, S.H.; Wilson, P.; Iyer, S.; Ferns, T.; Shapiro, R.; Brew, K.; Subramanian, V.; Acharya, K.R. Characterization of human angiogenin variants implicated in amyotrophic lateral sclerosis. Biochemistry 2007, 46, 11810–11818. [Google Scholar] [CrossRef] [PubMed]
- Nogues, M.V.; Vilanova, M.; Cuchillo, C.M. Bovine pancreatic ribonuclease A as a model of an enzyme with multiple substrate binding sites. Biochim. Biophys. Acta 1995, 1253, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yu, W.; Kishikawa, H.; Folkerth, R.D.; Iafrate, A.J.; Shen, Y.; Xin, W.; Sims, K.; Hu, G.F. Angiogenin loss-of-function mutations in amyotrophic lateral sclerosis. Ann. Neurol. 2007, 62, 609–617. [Google Scholar] [CrossRef]
- Gellera, C.; Colombrita, C.; Ticozzi, N.; Castellotti, B.; Bragato, C.; Ratti, A.; Taroni, F.; Silani, V. Identification of new ANG gene mutations in a large cohort of Italian patients with amyotrophic lateral sclerosis. Neurogenetics 2008, 9, 33–40. [Google Scholar] [CrossRef]
- Conforti, F.L.; Sprovieri, T.; Mazzei, R.; Ungaro, C.; La Bella, V.; Tessitore, A.; Patitucci, A.; Magariello, A.; Gabriele, A.L.; Tedeschi, G.; et al. A novel Angiogenin gene mutation in a sporadic patient with amyotrophic lateral sclerosis from southern Italy. Neuromuscul. Disord. 2008, 18, 68–70. [Google Scholar] [CrossRef]
- Paubel, A.; Violette, J.; Amy, M.; Praline, J.; Meininger, V.; Camu, W.; Corcia, P.; Andres, C.R.; Vourc’h, P.; French Amyotrophic Lateral Sclerosis Study, G. Mutations of the ANG gene in French patients with sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2008, 65, 1333–1336. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Seki, N.; Ishiura, H.; Mitsui, J.; Matsukawa, T.; Kishino, A.; Onodera, O.; Aoki, M.; Shimozawa, N.; Murayama, S.; et al. Development of a high-throughput microarray-based resequencing system for neurological disorders and its application to molecular genetics of amyotrophic lateral sclerosis. Arch. Neurol. 2008, 65, 1326–1332. [Google Scholar] [CrossRef]
- Fernandez-Santiago, R.; Hoenig, S.; Lichtner, P.; Sperfeld, A.D.; Sharma, M.; Berg, D.; Weichenrieder, O.; Illig, T.; Eger, K.; Meyer, T.; et al. Identification of novel Angiogenin (ANG) gene missense variants in German patients with amyotrophic lateral sclerosis. J. Neurol. 2009, 256, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, R.L.; Phukan, J.; McCormack, W.; Lynch, D.S.; Greenway, M.; Cronin, S.; Saunders, J.; Slowik, A.; Tomik, B.; Andersen, P.M.; et al. Angiogenin levels and ANG genotypes: Dysregulation in amyotrophic lateral sclerosis. PLoS ONE 2010, 5, e15402. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Min, J.; Staropoli, J.F.; Collin, E.; Bi, S.; Feng, X.; Barone, R.; Cao, Y.; O’Malley, L.; Xin, W.; et al. SOD1, ANG, TARDBP and FUS mutations in amyotrophic lateral sclerosis: A United States clinical testing lab experience. Amyotroph. Lateral Scler. 2012, 13, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.Y.; Wang, X.N.; Liu, M.S.; Sun, Q.; Li, X.G.; Cui, L.Y.; Kong, J. Identification of a novel missense mutation in angiogenin in a Chinese amyotrophic lateral sclerosis cohort. Amyotroph. Lateral Scler. 2012, 13, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Erriu, I.M.; Prehn, J.H. Molecular Mechanisms in Amyotrophic Lateral Sclerosis: The Role of Angiogenin, a Secreted RNase. Front. Neurosci. 2012, 6, 167. [Google Scholar] [CrossRef]
- Padhi, A.K.; Vasaikar, S.V.; Jayaram, B.; Gomes, J. Fast prediction of deleterious angiogenin mutations causing amyotrophic lateral sclerosis. FEBS Lett. 2013, 587, 1762–1766. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Kumar, H.; Vasaikar, S.V.; Jayaram, B.; Gomes, J. Mechanisms of loss of functions of human angiogenin variants implicated in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e32479. [Google Scholar] [CrossRef]
- Pous, J.; Canals, A.; Terzyan, S.S.; Guasch, A.; Benito, A.; Ribo, M.; Vilanova, M.; Coll, M. Three-dimensional structure of a human pancreatic ribonuclease variant, a step forward in the design of cytotoxic ribonucleases. J. Mol. Biol. 2000, 303, 49–60. [Google Scholar] [CrossRef]
- Padhi, A.K.; Narain, P.; Gomes, J. Rare Angiogenin and Ribonuclease 4 variants associated with amyotrophic lateral sclerosis exhibit loss-of-function: A comprehensive in silico study. Metab. Brain Dis. 2019, 34, 1661–1677. [Google Scholar] [CrossRef]
- Narain, P.; Padhi, A.K.; Dave, U.; Mishra, D.; Bhatia, R.; Vivekanandan, P.; Gomes, J. Identification and characterization of novel and rare susceptible variants in Indian amyotrophic lateral sclerosis patients. Neurogenetics 2019, 20, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Shukla, R.; Narain, P.; Gomes, J. A distant angiogenin variant causes amyotrophic lateral sclerosis through loss-of-function mechanisms: Insights from long-timescale atomistic simulations and conformational dynamics. Comput. Biol. Med. 2021, 135, 104602. [Google Scholar] [CrossRef] [PubMed]
- Tripolszki, K.; Danis, J.; Padhi, A.K.; Gomes, J.; Bozo, R.; Nagy, Z.F.; Nagy, D.; Klivenyi, P.; Engelhardt, J.I.; Szell, M. Angiogenin mutations in Hungarian patients with amyotrophic lateral sclerosis: Clinical, genetic, computational, and functional analyses. Brain Behav. 2019, 9, e01293. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, N.; Ferguson, R.; Subramanian, V.; Acharya, K.R. Structural and molecular insights into the mechanism of action of human angiogenin-ALS variants in neurons. Nat. Commun. 2012, 3, 1121. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, W.J.; Rehman, S.; Pham, T.T.; Thiyagarajan, N.; Lee, R.L.; Subramanian, V.; Acharya, K.R. Structural insights into human angiogenin variants implicated in Parkinson’s disease and Amyotrophic Lateral Sclerosis. Sci. Rep. 2017, 7, 41996. [Google Scholar] [CrossRef] [PubMed]
- Fagagnini, A.; Garavis, M.; Gomez-Pinto, I.; Fasoli, S.; Gotte, G.; Laurents, D.V. NMR Characterization of Angiogenin Variants and tRNA(Ala) Products Impacting Aberrant Protein Oligomerization. Int. J. Mol. Sci. 2021, 22, 1439. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Liu, Y.; Hart, P.J.; Schlunegger, M.P.; Eisenberg, D. The crystal structure of a 3D domain-swapped dimer of RNase A at a 2.1-A resolution. Proc. Natl. Acad. Sci. USA 1998, 95, 3437–3442. [Google Scholar] [CrossRef]
- Liu, Y.; Gotte, G.; Libonati, M.; Eisenberg, D. A domain-swapped RNase A dimer with implications for amyloid formation. Nat. Struct. Biol. 2001, 8, 211–214. [Google Scholar] [CrossRef]
- Gotte, G.; Bertoldi, M.; Libonati, M. Structural versatility of bovine ribonuclease A. Distinct conformers of trimeric and tetrameric aggregates of the enzyme. Eur. J. Biochem. 1999, 265, 680–687. [Google Scholar] [CrossRef]
- Liu, Y.; Gotte, G.; Libonati, M.; Eisenberg, D. Structures of the two 3D domain-swapped RNase A trimers. Protein Sci. 2002, 11, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Gotte, G.; Laurents, D.V.; Libonati, M. Three-dimensional domain-swapped oligomers of ribonuclease A: Identification of a fifth tetramer, pentamers and hexamers, and detection of trace heptameric, octameric and nonameric species. Biochim. Biophys. Acta 2006, 1764, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Eisenberg, D. 3D domain swapping: As domains continue to swap. Protein Sci. 2002, 11, 1285–1299. [Google Scholar] [CrossRef] [PubMed]
- Libonati, M.; Gotte, G. Oligomerization of bovine ribonuclease A: Structural and functional features of its multimers. Biochem. J. 2004, 380 Pt 2, 311–327. [Google Scholar] [CrossRef]
- Montioli, R.; Campagnari, R.; Fasoli, S.; Fagagnini, A.; Caloiu, A.; Smania, M.; Menegazzi, M.; Gotte, G. RNase A Domain-Swapped Dimers Produced Through Different Methods: Structure-Catalytic Properties and Antitumor Activity. Life 2021, 11, 168. [Google Scholar] [CrossRef] [PubMed]
- Gotte, G.; Laurents, D.V.; Merlino, A.; Picone, D.; Spadaccini, R. Structural and functional relationships of natural and artificial dimeric bovine ribonucleases: New scaffolds for potential antitumor drugs. FEBS Lett. 2013, 587, 3601–3608. [Google Scholar] [CrossRef]
- Teng, P.K.; Anderson, N.J.; Goldschmidt, L.; Sawaya, M.R.; Sambashivan, S.; Eisenberg, D. Ribonuclease A suggests how proteins self-chaperone against amyloid fiber formation. Protein Sci. 2012, 21, 26–37. [Google Scholar] [CrossRef]
- Sambashivan, S.; Liu, Y.; Sawaya, M.R.; Gingery, M.; Eisenberg, D. Amyloid-like fibrils of ribonuclease A with three-dimensional domain-swapped and native-like structure. Nature 2005, 437, 266–269. [Google Scholar] [CrossRef]
- Bennett, M.J.; Sawaya, M.R.; Eisenberg, D. Deposition diseases and 3D domain swapping. Structure 2006, 14, 811–824. [Google Scholar] [CrossRef]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef] [PubMed]
- Noji, M.; Samejima, T.; Yamaguchi, K.; So, M.; Yuzu, K.; Chatani, E.; Akazawa-Ogawa, Y.; Hagihara, Y.; Kawata, Y.; Ikenaka, K.; et al. Breakdown of supersaturation barrier links protein folding to amyloid formation. Commun. Biol. 2021, 4, 120. [Google Scholar] [CrossRef] [PubMed]
- Siraj, S.; Yameen, D.; Bhati, S.; Athar, T.; Khan, S.; Bhattacharya, J.; Islam, A.; Haque, M.M. Sugar osmolyte inhibits and attenuates the fibrillogenesis in RNase A: An in vitro and in silico characterizations. Int. J. Biol. Macromol. 2023, 253 Pt 8, 127378. [Google Scholar] [CrossRef]
- Ercole, C.; Lopez-Alonso, J.P.; Font, J.; Ribo, M.; Vilanova, M.; Picone, D.; Laurents, D.V. Crowding agents and osmolytes provide insight into the formation and dissociation of RNase A oligomers. Arch. Biochem. Biophys. 2011, 506, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Gotte, G.; Butturini, E.; Bettin, I.; Noro, I.; Mahmoud Helmy, A.; Fagagnini, A.; Cisterna, B.; Malatesta, M. Slow Evolution toward “Super-Aggregation” of the Oligomers Formed through the Swapping of RNase A N-Termini: A Wish for Amyloidosis? Int. J. Mol. Sci. 2022, 23, 11192. [Google Scholar] [CrossRef] [PubMed]
- Noro, I.; Bettin, I.; Fasoli, S.; Smania, M.; Lunardi, L.; Giannini, M.; Andreoni, L.; Montioli, R.; Gotte, G. Human RNase 1 can extensively oligomerize through 3D domain swapping thanks to the crucial contribution of its C-terminus. Int. J. Biol. Macromol. 2023, 249, 126110. [Google Scholar] [CrossRef]
- Canals, A.; Pous, J.; Guasch, A.; Benito, A.; Ribo, M.; Vilanova, M.; Coll, M. The structure of an engineered domain-swapped ribonuclease dimer and its implications for the evolution of proteins toward oligomerization. Structure 2001, 9, 967–976. [Google Scholar] [CrossRef]
- Pica, A.; Merlino, A.; Buell, A.K.; Knowles, T.P.; Pizzo, E.; D’Alessio, G.; Sica, F.; Mazzarella, L. Three-dimensional domain swapping and supramolecular protein assembly: Insights from the X-ray structure of a dimeric swapped variant of human pancreatic RNase. Acta Crystallogr. D Biol. Crystallogr. 2013, 69 Pt 10, 2116–2123. [Google Scholar] [CrossRef]
- Mazzarella, L.; Capasso, S.; Demasi, D.; Di Lorenzo, G.; Mattia, C.A.; Zagari, A. Bovine seminal ribonuclease: Structure at 1.9 A resolution. Acta Crystallogr. D Biol. Crystallogr. 1993, 49 Pt 4, 389–402. [Google Scholar] [CrossRef]
- Cafaro, V.; De Lorenzo, C.; Piccoli, R.; Bracale, A.; Mastronicola, M.R.; Di Donato, A.; D’Alessio, G. The antitumor action of seminal ribonuclease and its quaternary conformations. FEBS Lett. 1995, 359, 31–34. [Google Scholar] [CrossRef]
- Navarro, S.; Boix, E.; Cuchillo, C.M.; Nogues, M.V. Eosinophil-induced neurotoxicity: The role of eosinophil cationic protein/RNase 3. J. Neuroimmunol. 2010, 227, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Odorizzi, F.; Nogues, M.V.; Boix, E. Eosinophil cationic protein aggregation: Identification of an N-terminus amyloid prone region. Biomacromolecules 2010, 11, 1983–1990. [Google Scholar] [CrossRef] [PubMed]
- Mosimann, S.C.; Ardelt, W.; James, M.N. Refined 1.7 A X-ray crystallographic structure of P-30 protein, an amphibian ribonuclease with anti-tumor activity. J. Mol. Biol. 1994, 236, 1141–1153. [Google Scholar] [CrossRef]
- Fagagnini, A.; Pica, A.; Fasoli, S.; Montioli, R.; Donadelli, M.; Cordani, M.; Butturini, E.; Acquasaliente, L.; Picone, D.; Gotte, G. Onconase dimerization through 3D domain swapping: Structural investigations and increase in the apoptotic effect in cancer cells. Biochem. J. 2017, 474, 3767–3781. [Google Scholar] [CrossRef] [PubMed]
- Gotte, G.; Campagnari, R.; Loreto, D.; Bettin, I.; Calzetti, F.; Menegazzi, M.; Merlino, A. The crystal structure of the domain-swapped dimer of onconase highlights some catalytic and antitumor activity features of the enzyme. Int. J. Biol. Macromol. 2021, 191, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Sievers, K.; Ficner, R. Structure of angiogenin dimer bound to double-stranded RNA. Acta Crystallogr. F Struct. Biol. Commun. 2022, 78 Pt 9, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Chatzileontiadou, D.S.M.; Samiotaki, M.; Alexopoulou, A.N.; Cotsiki, M.; Panayotou, G.; Stamatiadi, M.; Balatsos, N.A.A.; Leonidas, D.D.; Kontou, M. Proteomic Analysis of Human Angiogenin Interactions Reveals Cytoplasmic PCNA as a Putative Binding Partner. J. Proteome Res. 2017, 16, 3606–3622. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, O.S.E.; Tsika, A.C.; Rovoli, M.; Papadopoulos, G.E.; Kontopidis, G.; Spyroulias, G.A.; Leonidas, D.D. Structural and Biochemical Characterization of the Human Angiogenin-Proliferating Cell Nuclear Antigen Interaction. Biochemistry 2023, 62, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sheng, J.; Hu, J.K.; Yu, W.; Kishikawa, H.; Hu, M.G.; Shima, K.; Wu, D.; Xu, Z.; Xin, W.; et al. Ribonuclease 4 protects neuron degeneration by promoting angiogenesis, neurogenesis, and neuronal survival under stress. Angiogenesis 2013, 16, 387–404. [Google Scholar] [CrossRef]
- Terzyan, S.S.; Peracaula, R.; de Llorens, R.; Tsushima, Y.; Yamada, H.; Seno, M.; Gomis-Ruth, F.X.; Coll, M. The three-dimensional structure of human RNase 4, unliganded and complexed with d(Up), reveals the basis for its uridine selectivity. J. Mol. Biol. 1999, 285, 205–214. [Google Scholar] [CrossRef]
- Rosenberg, H.F.; Dyer, K.D. Human ribonuclease 4 (RNase 4): Coding sequence, chromosomal localization and identification of two distinct transcripts in human somatic tissues. Nucleic Acids Res. 1995, 23, 4290–4295. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.; Bochter, M.S.; Simoni, A.; Bender, K.; de Dios Ruiz Rosado, J.; Cotzomi-Ortega, I.; Sanchez-Zamora, Y.I.; Becknell, B.; Linn, S.; Li, B.; et al. Repurposing HDAC inhibitors to enhance ribonuclease 4 and 7 expression and reduce urinary tract infection. Proc. Natl. Acad. Sci. USA 2023, 120, e2213363120. [Google Scholar] [CrossRef]
- Vanli, N.; Sheng, J.; Li, S.; Xu, Z.; Hu, G.F. Ribonuclease 4 is associated with aggressiveness and progression of prostate cancer. Commun. Biol. 2022, 5, 625. [Google Scholar] [CrossRef] [PubMed]
- Di Liddo, R.; Dalzoppo, D.; Baiguera, S.; Conconi, M.T.; Dettin, M.; Parnigotto, P.P.; Grandi, C. In vitro biological activity of bovine milk ribonuclease-4. Mol. Med. Rep. 2010, 3, 127–132. [Google Scholar] [PubMed]
- Padhi, A.K.; Narain, P.; Dave, U.; Satija, R.; Patir, A.; Gomes, J. Insights into the role of ribonuclease 4 polymorphisms in amyotrophic lateral sclerosis. J. Biomol. Struct. Dyn. 2019, 37, 116–130. [Google Scholar] [CrossRef]
- Padhi, A.K.; Gomes, J. A molecular dynamics based investigation reveals the role of rare Ribonuclease 4 variants in amyotrophic lateral sclerosis susceptibility. Mutat. Res. 2019, 813, 1–12. [Google Scholar] [CrossRef]
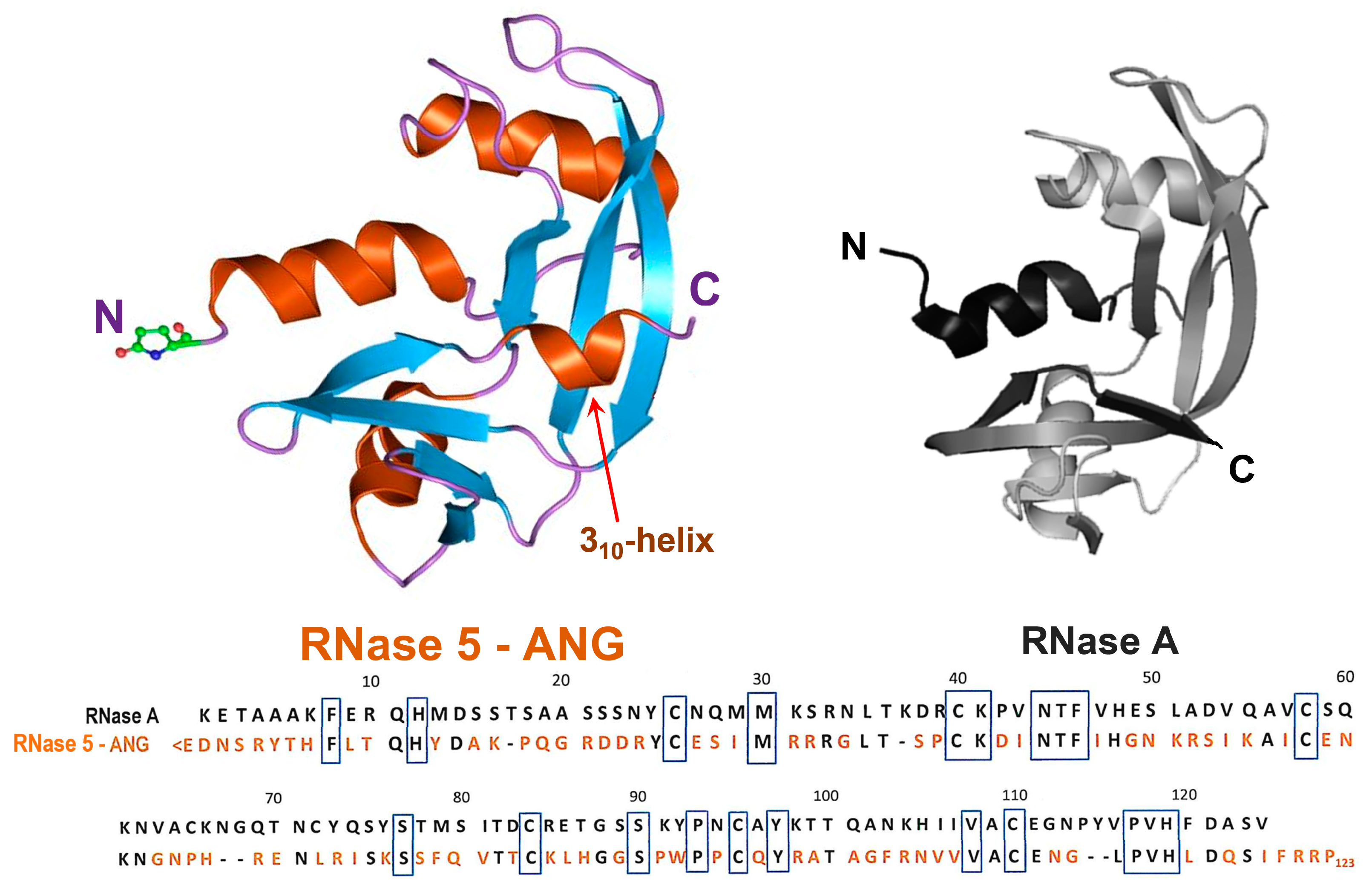
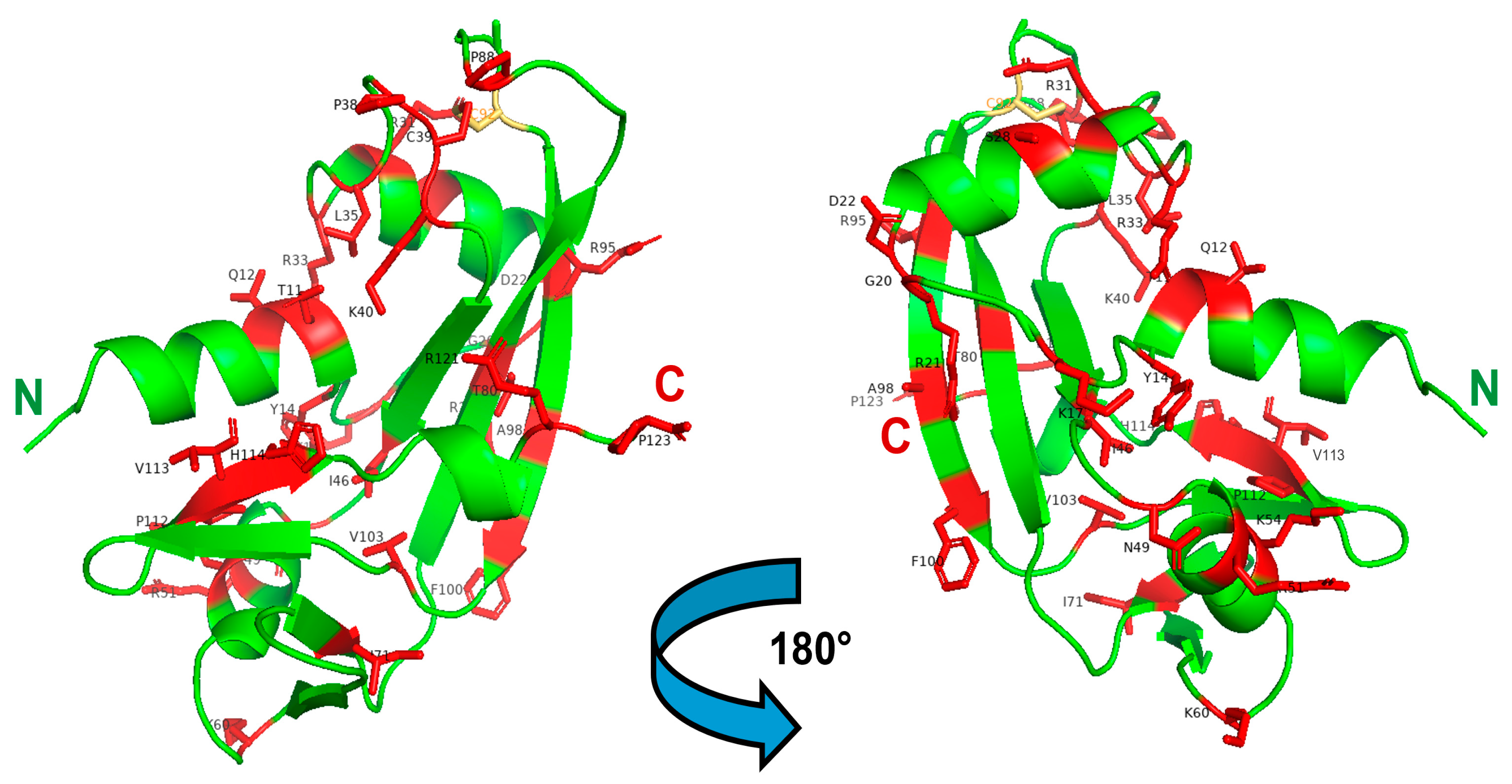
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).