A Subset of Microsatellite Unstable Cancer Genomes Prone to Short Insertions over Deletions Is Associated with Elevated Anticancer Immunity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Mutational Signature Analysis
2.3. Genes Differentially Targeted and Expressed between MMRd-ins and MMRd-del Genomes
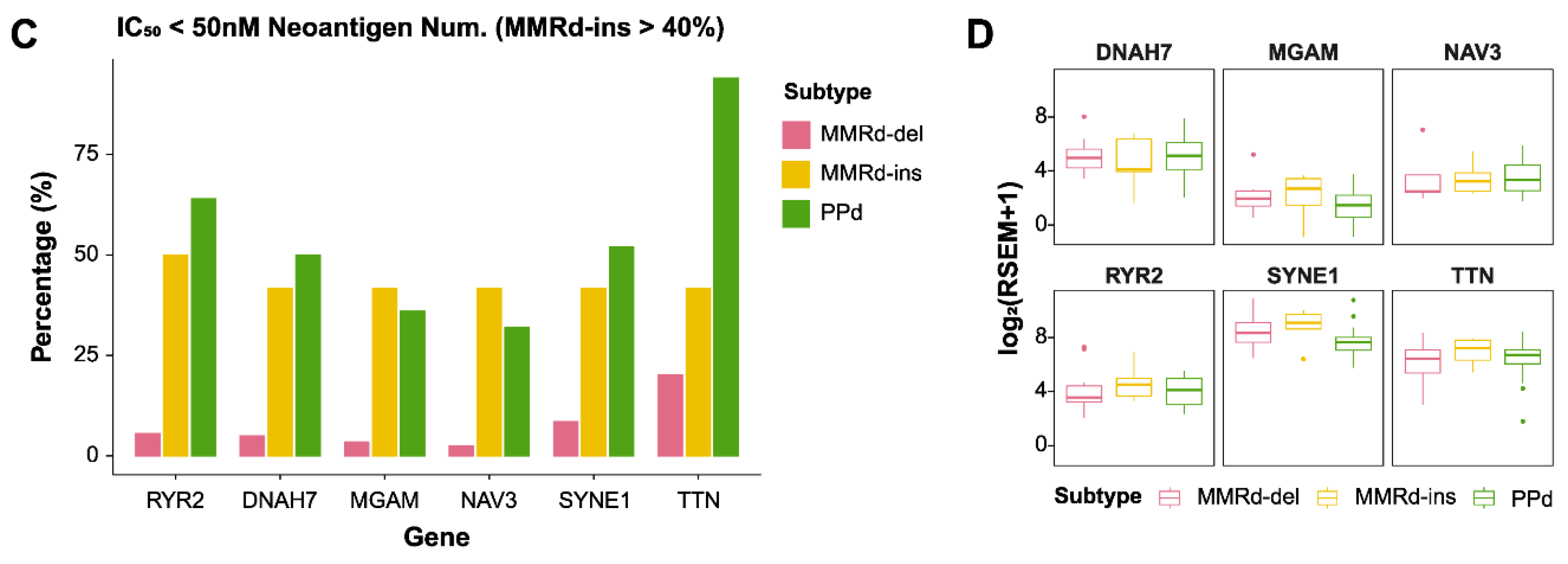
2.4. Identification of Neoantigens
2.5. Structural Variations
3. Results
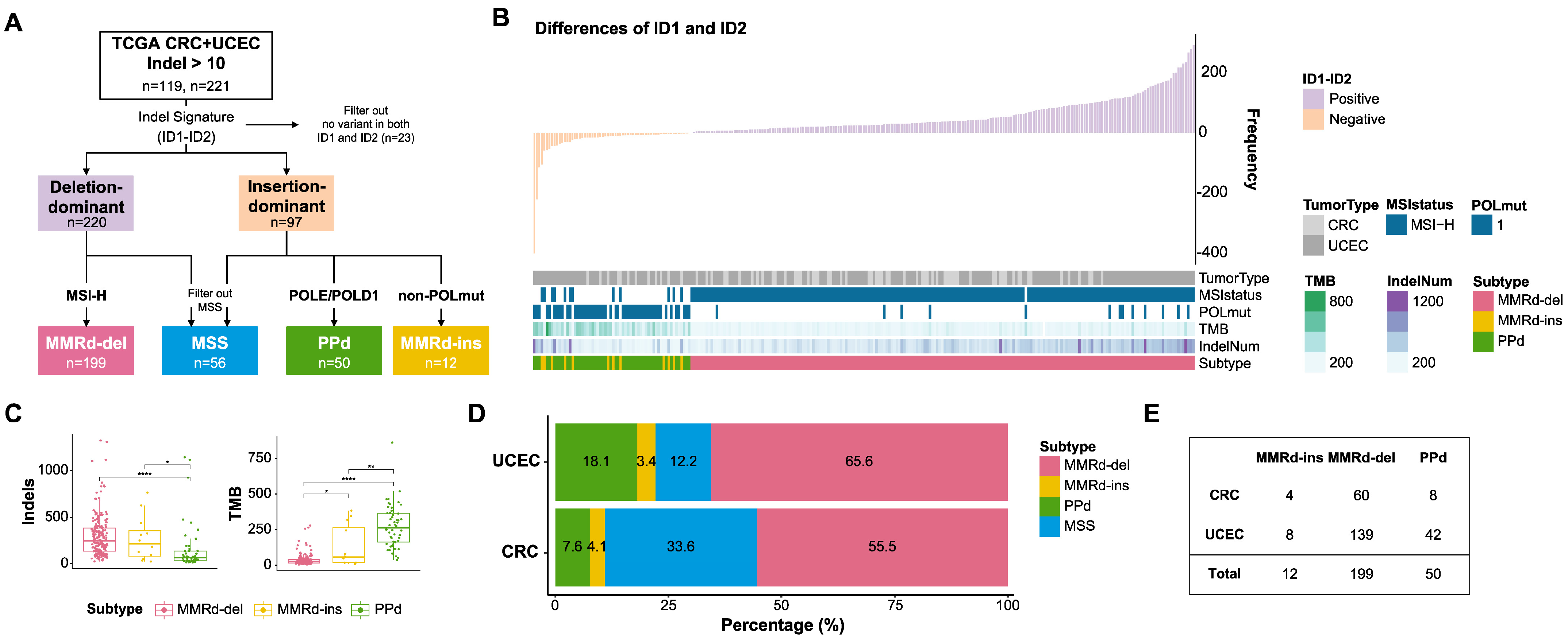
3.1. Indel-Based Classification of MMRd Genomes
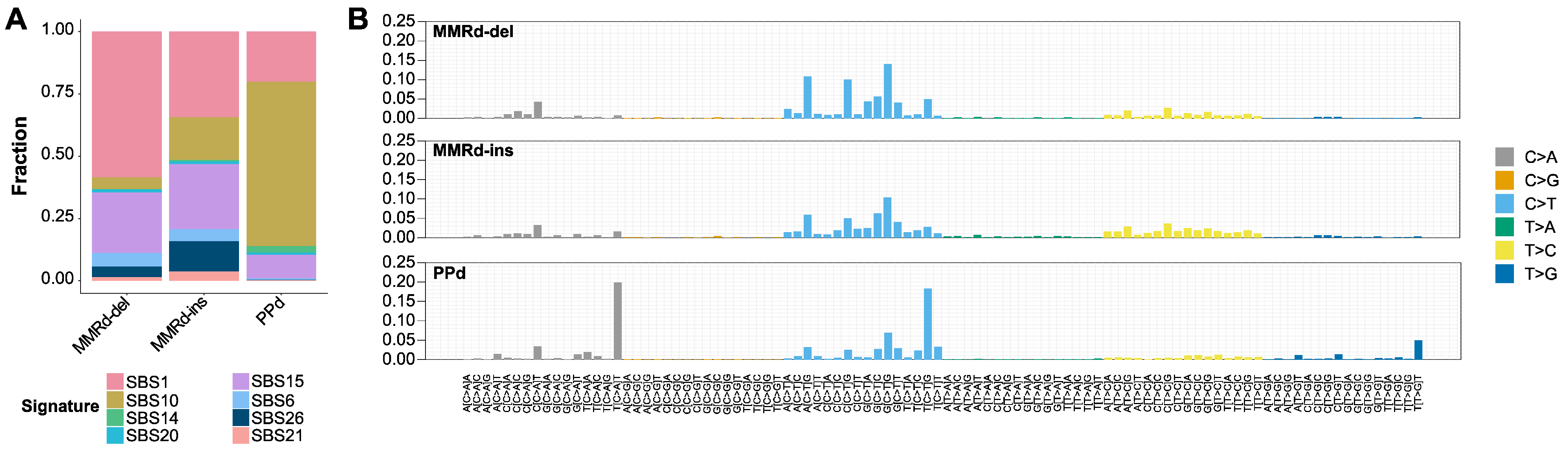
3.2. Mutational Signatures and Concordance between Subtypes
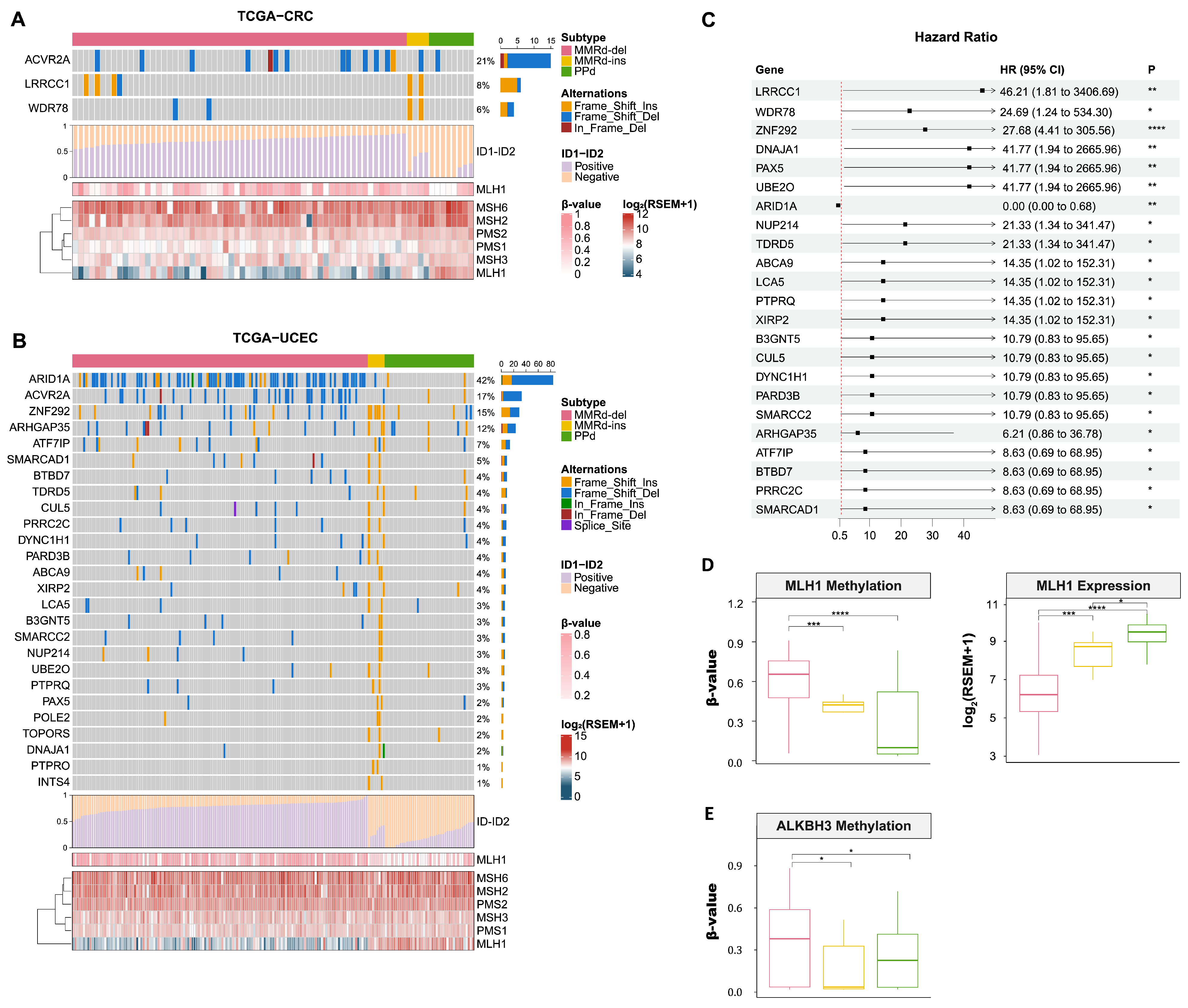
3.3. Mutational Landscape of MMRd-ins, MMRd-del, and PPd Genomes
3.4. Comparing Gene Expression between MMRd-del and MMRd-ins Genomes
3.5. Genomic Signatures of MMRd-del and -ins Genomes Based on Whole Genome Sequencing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chung, J.; Maruvka, Y.E.; Sudhaman, S.; Kelly, J.; Haradhvala, N.J.; Bianchi, V.; Edwards, M.; Forster, V.J.; Nunes, N.M.; Galati, M.A.; et al. DNA Polymerase and Mismatch Repair Exert Distinct Microsatellite Instability Signatures in Normal and Malignant Human Cells. Cancer Discov. 2021, 11, 1176–1191. [Google Scholar] [CrossRef]
- Lower, S.S.; McGurk, M.P.; Clark, A.G.; Barbash, D.A. Satellite DNA evolution: Old ideas, new approaches. Curr. Opin. Genet. Dev. 2018, 49, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, S.N.; Bren, G.; Schaid, D. Microsatellite Instability in Cancer of the Proximal Colon. Science 1993, 260, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.; Tsao, J.L.; Wu, A.; Shibata, D. Stepwise deletions of polyA sequences in mismatch repair-deficient colorectal cancers. Am. J. Pathol. 2001, 158, 1867–1870. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, Y.M.; de Jong, A.E.; Morreau, H.; Tops, C.M.; Vasen, H.F.; Wijnen, J.T.; Breuning, M.H.; Brocker-Vriends, A.H. Diagnostic approach and management of Lynch syndrome (hereditary nonpolyposis colorectal carcinoma): A guide for clinicians. CA Cancer J. Clin. 2006, 56, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Ciriano, I.; Lee, S.; Park, W.Y.; Kim, T.M.; Park, P.J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell International 2020, 20, 16. [Google Scholar] [CrossRef]
- Dudley, J.C.; Lin, M.-T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Gryfe, R.; Kim, H.; Hsieh, E.T.; Aronson, M.D.; Holowaty, E.J.; Bull, S.B.; Redston, M.; Gallinger, S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N. Engl. J. Med. 2000, 342, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Bilbao, C.; Lara, P.C.; Ramirez, R.; Henriquez-Hernandez, L.A.; Rodriguez, G.; Falcon, O.; Leon, L.; Perucho, M.; Diaz-Chico, B.N.; Diaz-Chico, J.C. Microsatellite instability predicts clinical outcome in radiation-treated endometrioid endometrial cancer. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Le, D.T. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 373, 1979. [Google Scholar] [CrossRef] [PubMed]
- Fader, A.N.; Diaz, L.A.; Armstrong, D.K.; Tanner, E.J.; Uram, J.; Eyring, A.; Wang, H.; Fisher, G.; Greten, T.; Le, D. Preliminary results of a phase II study: PD-1 blockade in mismatch repair–deficient, recurrent or persistent endometrial cancer. Gynecol. Oncol. 2016, 141, 206–207. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Kirchner, J.M.; Tran, H.; Resnick, M.A. A DNA polymerase epsilon mutant that specifically causes +1 frameshift mutations within homonucleotide runs in yeast. Genetics 2000, 155, 1623–1632. [Google Scholar] [CrossRef]
- Xing, X.; Kane, D.P.; Bulock, C.R.; Moore, E.A.; Sharma, S.; Chabes, A.; Shcherbakova, P.V. A recurrent cancer-associated substitution in DNA polymerase ε produces a hyperactive enzyme. Nat. Commun. 2019, 10, 374. [Google Scholar] [CrossRef]
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S.N.; et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-l.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370.e314. [Google Scholar] [CrossRef] [PubMed]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Haradhvala, N.J.; Kim, J.; Maruvka, Y.E.; Polak, P.; Rosebrock, D.; Livitz, D.; Hess, J.M.; Leshchiner, I.; Kamburov, A.; Mouw, K.W.; et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat. Commun. 2018, 9, 1746. [Google Scholar] [CrossRef] [PubMed]
- Hübschmann, D.; Jopp-Saile, L.; Andresen, C.; Krämer, S.; Gu, Z.; Heilig, C.E.; Kreutzfeldt, S.; Teleanu, V.; Fröhling, S.; Eils, R.; et al. Analysis of mutational signatures with yet another package for signature analysis. Genes Chromosomes Cancer 2021, 60, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. deconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- The Bioconductor Dev Team. BSgenome.Hsapiens.UCSC.hg19: Full Genome Sequences for Homo Sapiens (UCSC Version hg19); 2014. Available online: https://bioconductor.org/packages/release/data/annotation/html/BSgenome.Hsapiens.UCSC.hg19.html (accessed on 4 May 2023).
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Shi, Y.; Jing, B.; Xi, R. Comprehensive analysis of neoantigens derived from structural variation across whole genomes from 2528 tumors. Genome Biol. 2023, 24, 169. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Zhong, X.; Yang, Y.; Yang, L. Starfish infers signatures of complex genomic rearrangements across human cancers. Nat. Cancer 2022, 3, 1247–1259. [Google Scholar] [CrossRef]
- Allo, G.; Bernardini, M.Q.; Wu, R.C.; Shih Ie, M.; Kalloger, S.; Pollett, A.; Gilks, C.B.; Clarke, B.A. ARID1A loss correlates with mismatch repair deficiency and intact p53 expression in high-grade endometrial carcinomas. Mod. Pathol. 2014, 27, 255–261. [Google Scholar] [CrossRef]
- Guan, B.; Gao, M.; Wu, C.H.; Wang, T.L.; Shih Ie, M. Functional analysis of in-frame indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia 2012, 14, 986–993. [Google Scholar] [CrossRef]
- Hannah, J.; Zhou, P. Regulation of DNA damage response pathways by the cullin-RING ubiquitin ligases. DNA Repair 2009, 8, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Pandita, R.K.; Hambarde, S.; Mattoo, A.R.; Charaka, V.; Ahmed, K.M.; Iyer, S.P.; Hunt, C.R.; Pandita, T.K. SMARCAD1 Phosphorylation and Ubiquitination Are Required for Resection during DNA Double-Strand Break Repair. iScience 2018, 2, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Song, S.Y.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Frameshift mutations of a tumor suppressor gene ZNF292 in gastric and colorectal cancers with high microsatellite instability. Apmis 2016, 124, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Frameshift mutations of a chromatin-remodeling gene SMARCC2 in gastric and colorectal cancers with microsatellite instability. Apmis 2013, 121, 168–169. [Google Scholar] [CrossRef] [PubMed]
- Razin, A.; Cedar, H. DNA methylation and gene expression. Microbiol. Rev. 1991, 55, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Esteve-Puig, R.; Climent, F.; Piñeyro, D.; Domingo-Domènech, E.; Davalos, V.; Encuentra, M.; Rea, A.; Espejo-Herrera, N.; Soler, M.; Lopez, M.; et al. Epigenetic loss of m1A RNA demethylase ALKBH3 in Hodgkin lymphoma targets collagen, conferring poor clinical outcome. Blood 2021, 137, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, K.S.; Aska, E.-M.; Tuominen, M.M.; Kauppi, L. Tissue-specific reduction in MLH1 expression induces microsatellite instability in intestine of Mlh1+/− mice. DNA Repair 2021, 106, 103178. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.A.; Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Kandoth, C.; Dooling, D.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The Clonal Evolution of Tumor Cell Populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Artyomov, M.N.; Mardis, E.R.; Schreiber, R.D. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J. Clin. Investig. 2015, 125, 3413–3421. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Xiang, L.; Wang, R.; Xiong, Y.; Zhou, H.; Gu, H.; Wang, J.; Peng, L. Bioinformatic Analysis of Immune Significance of RYR2 Mutation in Breast Cancer. Biomed. Res. Int. 2021, 2021, 8072796. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Shen, Z.; Yang, T.; Wu, M. DNAH7 mutations benefit colorectal cancer patients receiving immune checkpoint inhibitors. Ann. Transl. Med. 2022, 10, 1335. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xiao, J.; Zhou, B.; Wei, J.; Luo, J.; Chen, W. SYNE1 mutation may enhance the response to immune checkpoint blockade therapy in clear cell renal cell carcinoma patients. Aging 2020, 12, 19316–19324. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Németh, E.; Lovrics, A.; Gervai, J.Z.; Seki, M.; Rospo, G.; Bardelli, A.; Szüts, D. Two main mutational processes operate in the absence of DNA mismatch repair. DNA Repair 2020, 89, 102827. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Levine, R.; Baylin, S.B.; Ellenson, L.H.; Herman, J.G. MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene 1998, 17, 2413–2417. [Google Scholar] [CrossRef]
- Simpkins, S.B.; Bocker, T.; Swisher, E.M.; Mutch, D.G.; Gersell, D.J.; Kovatich, A.J.; Palazzo, J.P.; Fishel, R.; Goodfellow, P.J. MLH1 promoter methylation and gene silencing is the primary cause of microsatellite instability in sporadic endometrial cancers. Hum. Mol. Genet. 1999, 8, 661–666. [Google Scholar] [CrossRef]
- Herman, J.G.; Umar, A.; Polyak, K.; Graff, J.R.; Ahuja, N.; Issa, J.-P.J.; Markowitz, S.; Willson, J.K.V.; Hamilton, S.R.; Kinzler, K.W.; et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 6870–6875. [Google Scholar] [CrossRef]
- Streisinger, G.; Okada, Y.; Emrich, J.; Newton, J.; Tsugita, A.; Terzaghi, E.; Inouye, M. Frameshift mutations and the genetic code. This paper is dedicated to Professor Theodosius Dobzhansky on the occasion of his 66th birthday. Cold Spring Harb. Symp. Quant. Biol. 1966, 31, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Ponting, C.P.; Copley, R.R. Occurrence and consequences of coding sequence insertions and deletions in Mammalian genomes. Genome Res. 2004, 14, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Koh, G.C.C.; Nanda, A.S.; Degasperi, A.; Urgo, K.; Roumeliotis, T.I.; Agu, C.A.; Badja, C.; Momen, S.; Young, J.; et al. A systematic CRISPR screen defines mutational mechanisms underpinning signatures caused by replication errors and endogenous DNA damage. Nat. Cancer 2021, 2, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, B.; Lee, A.; Cabrera, B.L.; Miyai, K.; Luo, L.; Ramamoorthy, S.; Keku, T.; Sandler, R.S.; McGuire, K.L.; Carethers, J.M. Relationship of EMAST and microsatellite instability among patients with rectal cancer. J. Gastrointest. Surg. 2010, 14, 1521–1528. [Google Scholar] [CrossRef]
- Kondelin, J.; Martin, S.; Katainen, R.; Renkonen-Sinisalo, L.; Lepistö, A.; Koskensalo, S.; Böhm, J.; Mecklin, J.-P.; Cajuso, T.; Hänninen, U.A.; et al. No evidence of EMAST in whole genome sequencing data from 248 colorectal cancers. Genes Chromosomes Cancer 2021, 60, 463–473. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Han, D.-J.; Lee, S.-Y.; Moon, Y.; Kang, S.J.; Kim, T.-M. A Subset of Microsatellite Unstable Cancer Genomes Prone to Short Insertions over Deletions Is Associated with Elevated Anticancer Immunity. Genes 2024, 15, 770. https://doi.org/10.3390/genes15060770
Kim S, Han D-J, Lee S-Y, Moon Y, Kang SJ, Kim T-M. A Subset of Microsatellite Unstable Cancer Genomes Prone to Short Insertions over Deletions Is Associated with Elevated Anticancer Immunity. Genes. 2024; 15(6):770. https://doi.org/10.3390/genes15060770
Chicago/Turabian StyleKim, Sunmin, Dong-Jin Han, Seo-Young Lee, Youngbeen Moon, Su Jung Kang, and Tae-Min Kim. 2024. "A Subset of Microsatellite Unstable Cancer Genomes Prone to Short Insertions over Deletions Is Associated with Elevated Anticancer Immunity" Genes 15, no. 6: 770. https://doi.org/10.3390/genes15060770