
A Proteogenomic Approach to Unravel New Proteins Encoded in the Leishmania donovani (HU3) Genome

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
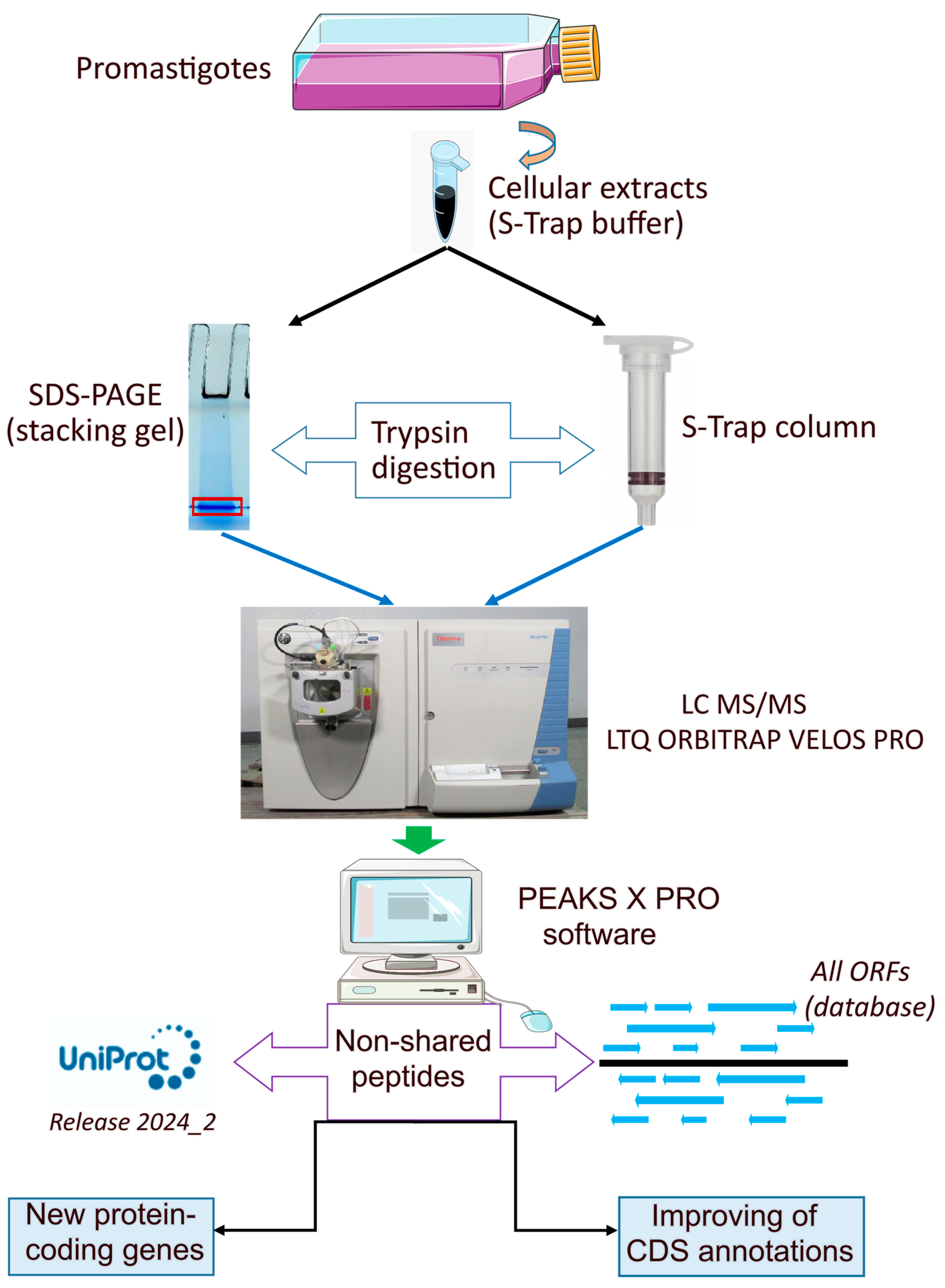
2.1. Parasite Culture and Preparations of Samples
2.2. Preparation of Protein Extract in STRAP Buffer and Digestion in Column (S-Trap Mini)
2.3. In-Gel Digestion
2.4. Reverse Phase-Liquid Chromatography (RP-LC)-MS/MS Analysis (Dynamic Exclusion Mode)
2.5. Data Analysis
2.6. Data Availability
3. Results
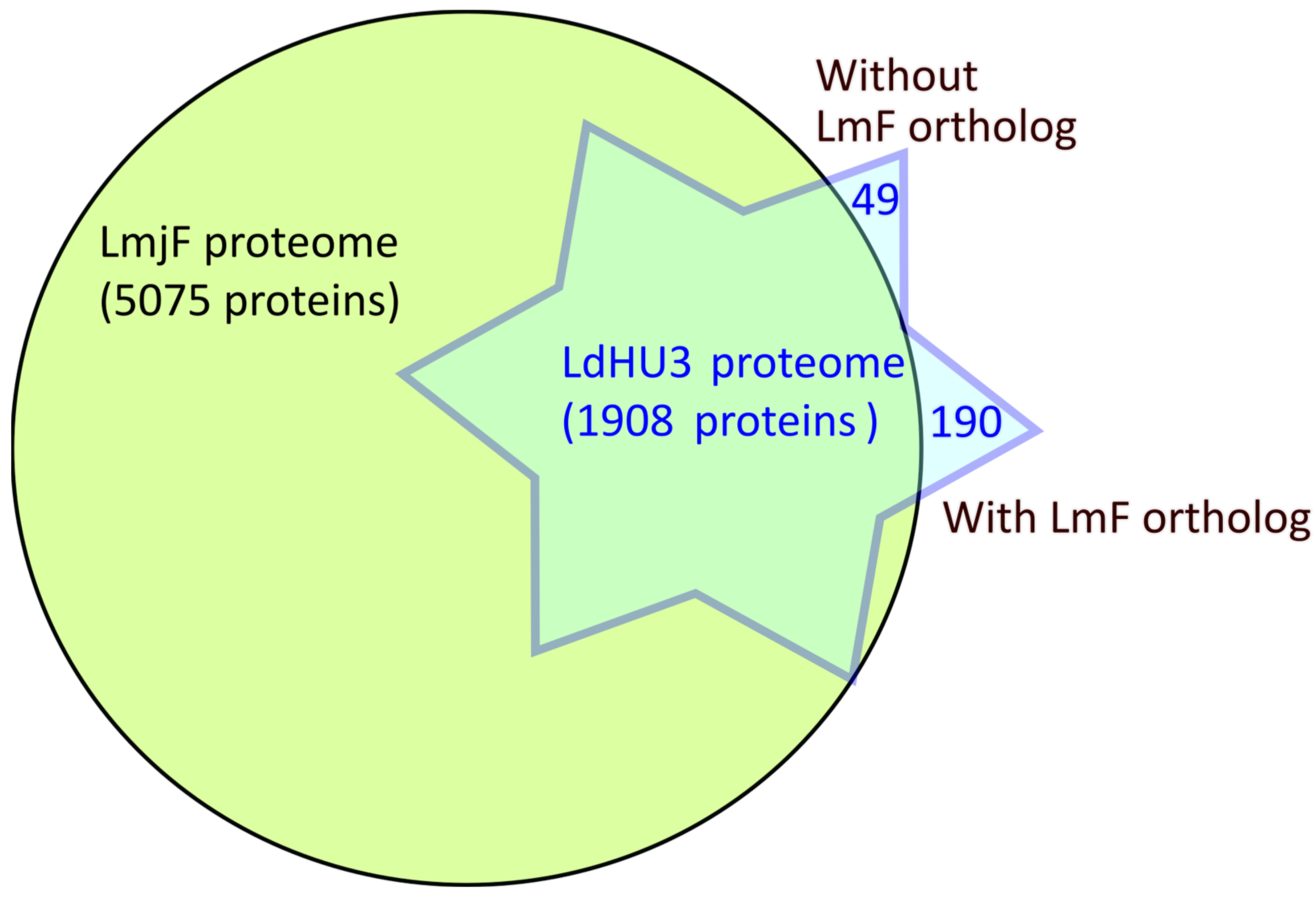
3.1. L. donovani (HU3) Experimental Proteome Determined by Protein Identification from LC−MS/MS Peptide Spectra
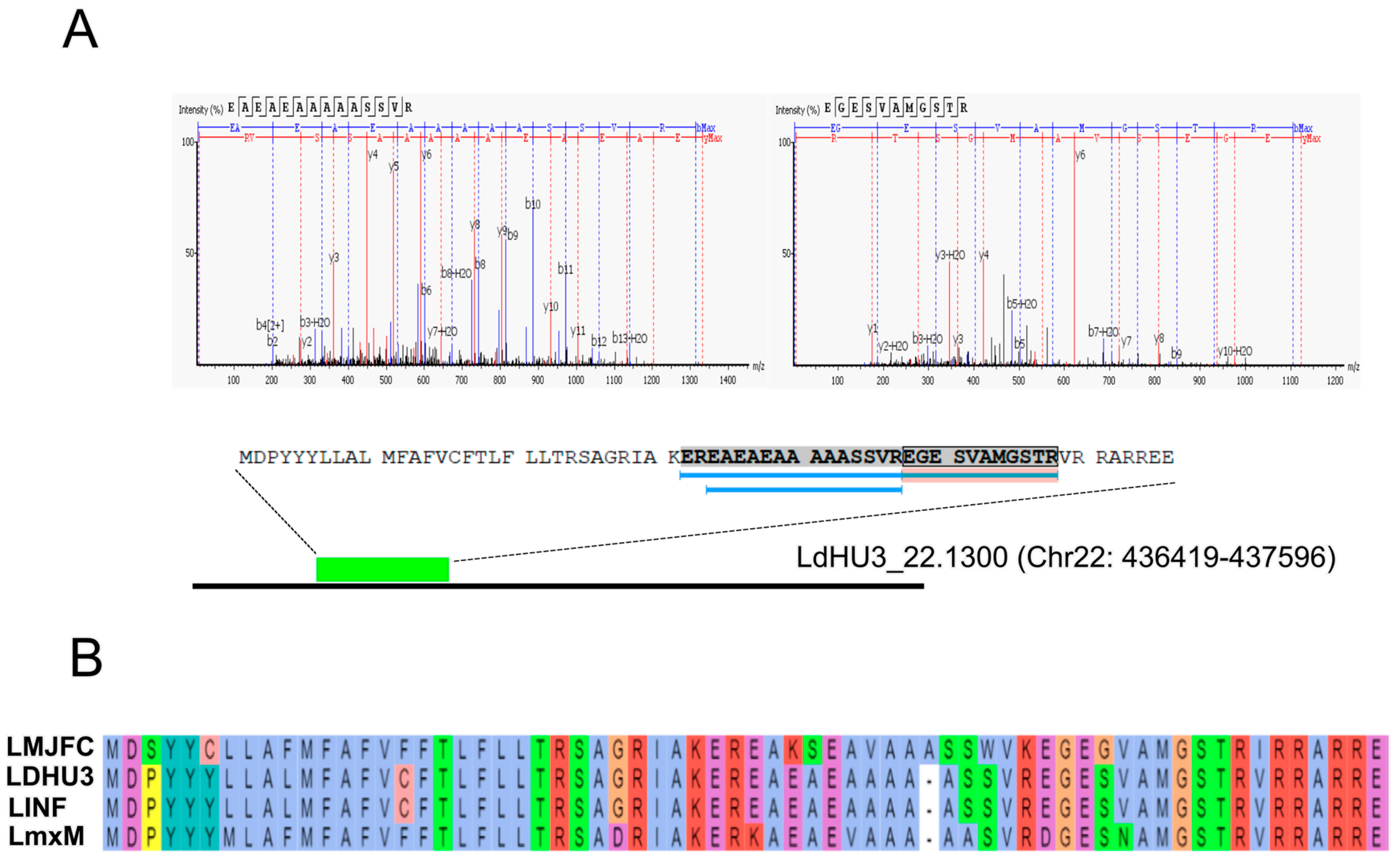
3.2. Annotation of New Protein-Coding Genes in the L. donovani (HU3) Genome
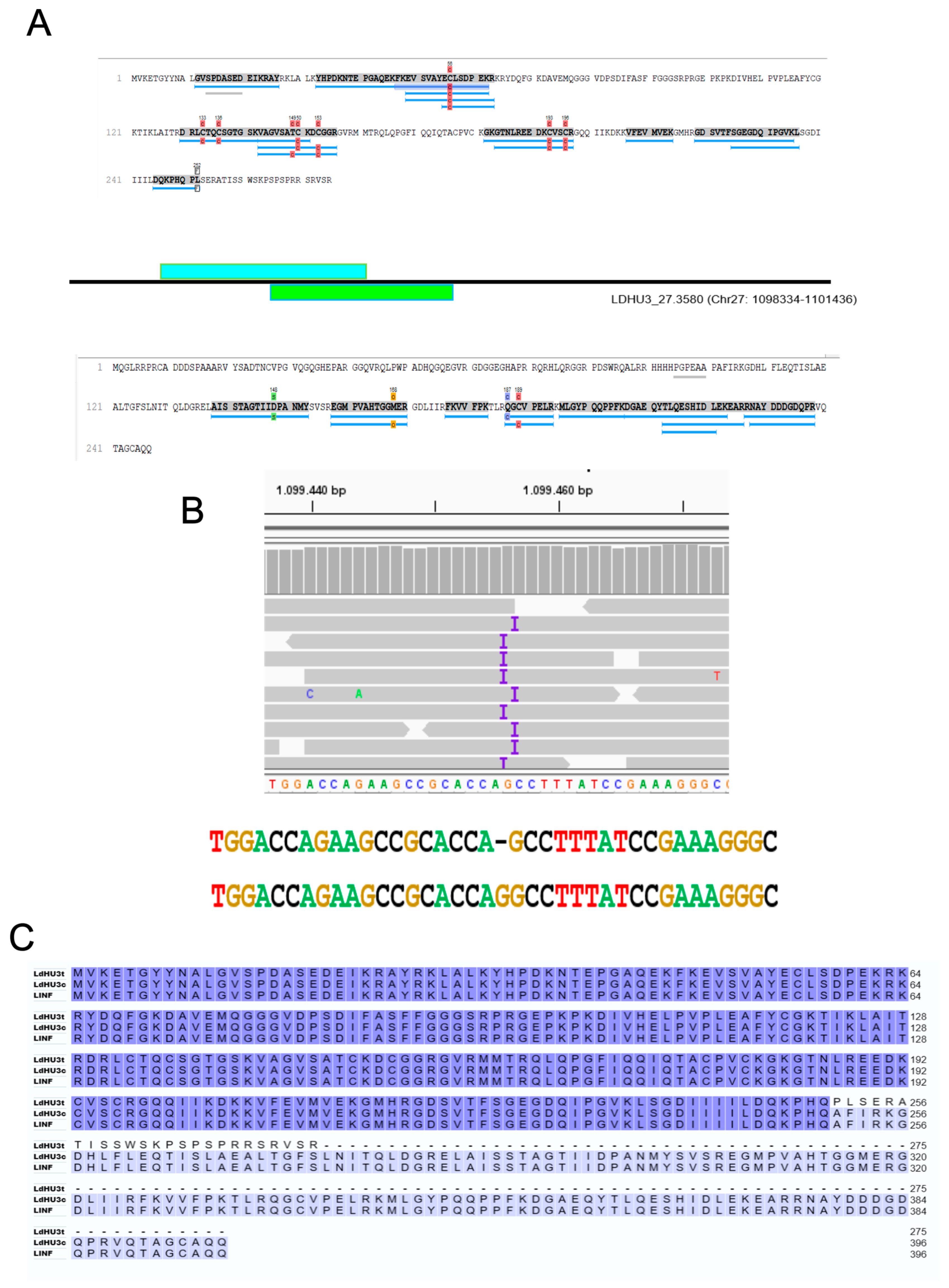
3.3. Re-Annotation of Gene Coding Sequences (CDS) to Accommodate Peptides Identified by the Proteomics Data
3.4. Identification of Two InDels in the Assembled L. dononani Genome Based on Proteomics Data
3.5. Identification of Post-Translational Modifications (PTMs) in L. donovani Proteins
3.6. Active Curation of L. donovani (HU3) Gene Annotations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alvar, J.; Velez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Solana, J.C.; Moreno, J.; Iborra, S.; Soto, M.; Requena, J.M. Live attenuated vaccines, a favorable strategy to provide long-term immunity against protozoan diseases. Trends Parasitol. 2022, 38, 316–334. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Singh, A. Chemotherapeutics of visceral leishmaniasis: Present and future developments. Parasitology 2018, 145, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Volpedo, G.; Huston, R.H.; Holcomb, E.A.; Pacheco-Fernandez, T.; Gannavaram, S.; Bhattacharya, P.; Nakhasi, H.L.; Satoskar, A.R. From infection to vaccination: Reviewing the global burden, history of vaccine development, and recurring challenges in global leishmaniasis protection. Expert. Rev. Vaccines 2021, 20, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Kumari, I.; Lakhanpal, D.; Swargam, S.; Nath Jha, A. Leishmaniasis: Omics Approaches to Understand its Biology from Molecule to Cell Level. Curr. Protein Pept. Sci. 2023, 24, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votypka, J.; Marty, P.; Delaunay, P.; Sereno, D. A Historical Overview of the Classification, Evolution, and Dispersion of Leishmania Parasites and Sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349. [Google Scholar] [CrossRef]
- Gupta, A.K.; Das, S.; Kamran, M.; Ejazi, S.A.; Ali, N. The pathogenicity and virulence of Leishmania—Interplay of virulence factors with host defenses. Virulence 2022, 13, 903–935. [Google Scholar] [CrossRef]
- Requena, J.M.; Alcolea, P.J.; Alonso, A.; Larraga, V. Omics approaches for understanding gene expression in Leishmania: Clues for tackling leishmaniasis. In Protozoan Parasitism—From Omics to Prevention and Control; Pablos-Torró, L.M., Lorenzo-Morales, J., Eds.; Caister Academic Press: Poole, UK, 2018; Chapter 5; pp. 77–112. [Google Scholar] [CrossRef]
- Cuervo, P.; Domont, G.B.; De Jesus, J.B. Proteomics of trypanosomatids of human medical importance. J. Proteom. 2010, 73, 845–867. [Google Scholar] [CrossRef] [PubMed]
- Paape, D.; Barrios-Llerena, M.E.; Le Bihan, T.; Mackay, L.; Aebischer, T. Gel free analysis of the proteome of intracellular Leishmania mexicana. Mol. Biochem. Parasitol. 2010, 169, 108–114. [Google Scholar] [CrossRef]
- Beneke, T.; Demay, F.; Hookway, E.; Ashman, N.; Jeffery, H.; Smith, J.; Valli, J.; Becvar, T.; Myskova, J.; Lestinova, T.; et al. Genetic dissection of a Leishmania flagellar proteome demonstrates requirement for directional motility in sand fly infections. PLoS Pathog. 2019, 15, e1007828. [Google Scholar] [CrossRef]
- McCall, L.I.; Zhang, W.W.; Dejgaard, K.; Atayde, V.D.; Mazur, A.; Ranasinghe, S.; Liu, J.; Olivier, M.; Nilsson, T.; Matlashewski, G. Adaptation of Leishmania donovani to cutaneous and visceral environments: In vivo selection and proteomic analysis. J. Proteome Res. 2015, 14, 1033–1059. [Google Scholar] [CrossRef] [PubMed]
- Tasbihi, M.; Shekari, F.; Hajjaran, H.; Masoori, L.; Hadighi, R. Mitochondrial proteome profiling of Leishmania tropica. Microb. Pathog. 2019, 133, 103542. [Google Scholar] [CrossRef]
- Jardim, A.; Hardie, D.B.; Boitz, J.; Borchers, C.H. Proteomic Profiling of Leishmania donovani Promastigote Subcellular Organelles. J. Proteome Res. 2018, 17, 1194–1215. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, D.; Smith, D.; Opperdoes, F.; Stern, S.; Olafson, R.W.; Zilberstein, D. Retooling Leishmania metabolism: From sand fly gut to human macrophage. Faseb. J. 2008, 22, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Sanchiz, Á.; Morato, E.; Rastrojo, A.; Camacho, E.; González-de la Fuente, S.; Marina, A.; Aguado, B.; Requena, J.M. The Experimental Proteome of Leishmania infantum Promastigote and Its Usefulness for Improving Gene Annotations. Genes 2020, 11, 1036. [Google Scholar] [CrossRef] [PubMed]
- Polanco, G.; Scott, N.E.; Lye, L.F.; Beverley, S.M. Expanded Proteomic Survey of the Human Parasite Leishmania major Focusing on Changes in Null Mutants of the Golgi GDP-Mannose/Fucose/Arabinopyranose Transporter LPG2 and of the Mitochondrial Fucosyltransferase FUT1. Microbiol. Spectr. 2022, 10, e0305222. [Google Scholar] [CrossRef] [PubMed]
- Pinho, N.; Wiśniewski, J.R.; Dias-Lopes, G.; Saboia-Vahia, L.; Bombaça, A.C.S.; Mesquita-Rodrigues, C.; Menna-Barreto, R.; Cupolillo, E.; de Jesus, J.B.; Padrón, G.; et al. In-depth quantitative proteomics uncovers specie-specific metabolic programs in Leishmania (Viannia) species. PLoS Negl. Trop. Dis. 2020, 14, e0008509. [Google Scholar] [CrossRef]
- Erben, E.D. High-throughput Methods for Dissection of Trypanosome Gene Regulatory Networks. Curr. Genom. 2018, 19, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Tran, N.H.; Qiao, R.; Xin, L.; Chen, X.; Liu, C.; Zhang, X.; Shan, B.; Ghodsi, A.; Li, M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat. Methods 2019, 16, 63–66. [Google Scholar] [CrossRef]
- Camacho, E.; González-de la Fuente, S.; Rastrojo, A.; Peiró-Pastor, R.; Solana, J.C.; Tabera, L.; Gamarro, F.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Complete assembly of the Leishmania donovani (HU3 strain) genome and transcriptome annotation. Sci. Rep. 2019, 9, 6127. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.F.; Peacock, C.S.; Cruz, A.K. Comparative genomics: From genotype to disease phenotype in the leishmaniases. Int. J. Parasitol. 2007, 37, 1173–1186. [Google Scholar] [CrossRef]
- Ivens, A.C.; Peacock, C.S.; Worthey, E.A.; Murphy, L.; Aggarwal, G.; Berriman, M.; Sisk, E.; Rajandream, M.A.; Adlem, E.; Aert, R.; et al. The Genome of the Kinetoplastid Parasite, Leishmania major. Science 2005, 309, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Camacho, E.; González-de la Fuente, S.; Solana, J.C.; Rastrojo, A.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Gene annotation and transcriptome delineation on a de novo genome assembly for the reference Leishmania major Friedlin strain. Genes 2021, 12, 1359. [Google Scholar] [CrossRef]
- Sánchez-Salvador, A.; González-de la Fuente, S.; Aguado, B.; Yates, P.A.; Requena, J.M. Refinement of Leishmania donovani Genome Annotations in the Light of Ribosome-Protected mRNAs Fragments (Ribo-Seq Data). Genes 2023, 14, 1637. [Google Scholar] [CrossRef] [PubMed]
- Pawar, H.; Pai, K.; Patole, M.S. A novel protein coding potential of long intergenic non-coding RNAs (lincRNAs) in the kinetoplastid protozoan parasite Leishmania major. Acta Trop. 2017, 167, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef] [PubMed]
- Nepomuceno-Mejía, T.; Florencio-Martínez, L.E.; Pineda-García, I.; Martínez-Calvillo, S. Identification of factors involved in ribosome assembly in the protozoan parasite Leishmania major. Acta Trop. 2022, 228, 106315. [Google Scholar] [CrossRef]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Br. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Manzano-Román, R.; Fuentes, M. Relevance and proteomics challenge of functional posttranslational modifications in Kinetoplastid parasites. J. Proteom. 2020, 220, 103762. [Google Scholar] [CrossRef]
- Halliday, C.; de Castro-Neto, A.; Alcantara, C.L.; Cunha-e-Silva, N.L.; Vaughan, S.; Sunter, J.D. Trypanosomatid Flagellar Pocket from Structure to Function. Trends Parasitol. 2021, 37, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Boucher, N.; Dacheux, D.; Giroud, C.; Baltz, T. An essential cell cycle-regulated nucleolar protein relocates to the mitotic spindle where it is involved in mitotic progression in Trypanosoma brucei. J. Biol. Chem. 2007, 282, 13780–13790. [Google Scholar] [CrossRef] [PubMed]
- Kushawaha, P.K.; Pati Tripathi, C.D.; Dube, A. Leishmania donovani secretory protein nucleoside diphosphate kinase b localizes in its nucleus and prevents ATP mediated cytolysis of macrophages. Microb. Pathog. 2022, 166, 105457. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.D.; Vieira, D.P.; Sanchez, M.A.; Valli, J.; Gluenz, E.; Landfear, S.M. Kharon1 null mutants of Leishmania mexicana are avirulent in mice and exhibit a cytokinesis defect within macrophages. PLoS ONE 2015, 10, e0134432. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Marmorstein, R. Protein N-terminal Acetylation: Structural Basis, Mechanism, Versatility, and Regulation. Trends Biochem. Sci. 2021, 46, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Aksnes, H.; McTiernan, N.; Arnesen, T. NATs at a glance. J. Cell Sci. 2023, 136, jcs260766. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Val, A.; Guzmán, U.H.; Olsen, J.V. Obtaining Complete Human Proteomes. Annu. Rev. Genom. Hum. Genet. 2022, 23, 99–121. [Google Scholar] [CrossRef] [PubMed]
- Flohe, L.; Budde, H.; Bruns, K.; Castro, H.; Clos, J.; Hofmann, B.; Kansal-Kalavar, S.; Krumme, D.; Menge, U.; Plank-Schumacher, K.; et al. Tryparedoxin peroxidase of Leishmania donovani: Molecular cloning, heterologous expression, specificity, and catalytic mechanism. Arch. Biochem. Biophys. 2002, 397, 324–335. [Google Scholar] [CrossRef]
- Kursula, I.; Wierenga, R.K. Crystal structure of triosephosphate isomerase complexed with 2-phosphoglycolate at 0.83-Å resolution. J. Biol. Chem. 2003, 278, 9544–9551. [Google Scholar] [CrossRef]
- Mukherjee, S.; Hao, Y.H.; Orth, K. A newly discovered post-translational modification--the acetylation of serine and threonine residues. Trends Biochem. Sci. 2007, 32, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Assis, L.A.; Santos Filho, M.V.C.; da Cruz Silva, J.R.; Bezerra, M.J.R.; de Aquino, I.R.P.U.C.; Merlo, K.C.; Holetz, F.B.; Probst, C.M.; Rezende, A.M.; Papadopoulou, B.; et al. Identification of novel proteins and mRNAs differentially bound to the Leishmania Poly(A) Binding Proteins reveals a direct association between PABP1, the RNA-binding protein RBP23 and mRNAs encoding ribosomal proteins. PLoS Negl. Trop. Dis. 2021, 15, e0009899. [Google Scholar] [CrossRef] [PubMed]
- Colineau, L.; Clos, J.; Moon, K.M.; Foster, L.J.; Reiner, N.E. Leishmania donovani chaperonin 10 regulates parasite internalization and intracellular survival in human macrophages. Med. Microbiol. Immunol. 2017, 206, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Requena, J.M.; Montalvo, A.M.; Fraga, J. Molecular Chaperones of Leishmania: Central Players in Many Stress-Related and -Unrelated Physiological Processes. Biomed. Res. Int. 2015, 2015, 301326. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.R.; Jensen, B.C.; Sur, A.; Wong, I.L.K.; Beverley, S.M.; Myler, P.J. Localization of Epigenetic Markers in Leishmania Chromatin. Pathogens 2022, 11, 930. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, L.; Quilles, J.C.; Campagnaro, G.D.; Azevedo Orsine, L.; Almeida, L.; Veras, F.; Miserani Magalhães, R.D.; Alcoforado Diniz, J.; Rodrigues Ferreira, T.; Cruz, A.K. Functional Study of Leishmania braziliensis Protein Arginine Methyltransferases (PRMTs) Reveals That PRMT1 and PRMT5 Are Required for Macrophage Infection. ACS Infect. Dis. 2022, 8, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Sprung, R.; Chen, Y.; Zhang, K.; Cheng, D.; Zhang, T.; Peng, J.; Zhao, Y. Identification and validation of eukaryotic aspartate and glutamate methylation in proteins. J. Proteome Res. 2008, 7, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, D.; Smith, D.; Myler, P.J.; Olafson, R.W.; Zilberstein, D. Post-translational modification of cellular proteins during Leishmania donovani differentiation. Proteomics 2008, 8, 1843–1850. [Google Scholar] [CrossRef]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. Engl. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Nirujogi, R.S.; Pawar, H.; Renuse, S.; Kumar, P.; Chavan, S.; Sathe, G.; Sharma, J.; Khobragade, S.; Pande, J.; Modak, B.; et al. Moving from unsequenced to sequenced genome: Reanalysis of the proteome of Leishmania donovani. J. Proteom. 2014, 97, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Hem, S.; Gherardini, P.F.; Osorio y Fortea, J.; Hourdel, V.; Morales, M.A.; Watanabe, R.; Pescher, P.; Kuzyk, M.A.; Smith, D.; Borchers, C.H.; et al. Identification of Leishmania-specific protein phosphorylation sites by LC-ESI-MS/MS and comparative genomics analyses. Proteomics 2010, 10, 3868–3883. [Google Scholar] [CrossRef] [PubMed]
- Tsigankov, P.; Gherardini, P.F.; Helmer-Citterich, M.; Spath, G.F.; Myler, P.J.; Zilberstein, D. Regulation dynamics of Leishmania differentiation: Deconvoluting signals and identifying phosphorylation trends. Mol. Cell Proteom. 2014, 13, 1787–1799. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Mass (Da) | #Peptides | #Unique | Product |
|---|---|---|---|---|
| LDHU3_02.0870 | 17,391 | 3 | 1 | Peptidase M3A/M3B family member |
| LDHU3_05.1170 | 50,820 | 5 | 5 | Protein of unknown function |
| LDHU3_08.0490 | 124,532 | 4 | 4 | Protein of unknown function |
| LDHU3_11.1460 | 208,953 | 3 | 3 | ATP-binding cassette subfamily A, member 1 |
| LDHU3_11.1500 | 208,645 | 3 | 3 | ATP-binding cassette protein subfamily A, member 4 |
| LDHU3_11.1540 | 208,645 | 3 | 3 | ATP-binding cassette protein subfamily A, member 4 |
| LDHU3_20.1640 | 17,036 | 6 | 6 | Small myristoylated protein 4 |
| LDHU3_22.1300 | 7394 | 3 | 3 | Protein of unknown function |
| LDHU3_27.0640 | 654,271 | 62 | 59 | Calpain-like cysteine peptidase |
| LDHU3_29.3160 | 69,485 | 7 | 7 | Domain of unknown function (DUF4139) |
| LDHU3_29.3180 | 195,556 | 2 | 2 | UDP-glucose/Glycoprotein Glucosyltransferase |
| LDHU3_30.5010 | 11,969 | 2 | 2 | Protein of unknown function |
| LDHU3_32.4380 | 58,558 | 21 | 21 | T-complex protein 1 subunit α|TCP1α|CCT-alfa |
| LDHU3_32.4600 | 9779 | 2 | 2 | Protein of unknown function |
| LDHU3_33.4490 | 132,996 | 6 | 6 | Protein of unknown function |
| LDHU3_34.1180 | 185,525 | 6 | 2 | Flagellar attachment zone protein |
| LDHU3_34.1190 | 281,656 | 9 | 5 | Flagellar attachment zone protein|FAZ1 |
| LDHU3_35.0470 | 43,405 | 12 | 11 | ATP-dependent DEAD-box RNA helicase|DHH1 |
| LDHU3_35.6550 | 90,643 | 6 | 6 | Zinc finger protein family member|ZC3H28 |
| LDHU3_36.7950 | 5153 | 3 | 3 | Protein of unknown function |
| Functional Category | Proteins * |
|---|---|
| Ribosomal proteins | eIF4A1, uL16, eL8, eL40, EF1G, eS21, uS8, eS12, eS4, uL1, uS15, eS6, uS19, uL11, uL29, eS26, uS11, RACK1, eL13, eL40, uL3, eEF1Bβ, uL3, eS1, eEF2, uS13, eS10, eEF1Bα, L10a |
| Protein folding | HOP, Aha1, HSP100, HSP110, HSP70.4, CCT-β, GRP78, HSP70, mtHSP70, HSP83/90, TRAP-1, HOP2, HSP60, Cyp19 |
| RNA-binding proteins | TSR1, SNU13, RBP42, HEL67, DRBD18, ALBA3, DRBD2, RNA helicase, PABP2, PUF11, ribonucleoprotein p18, L-PSP |
| Oxidative stress | Thioredoxin, tryparedoxin peroxidase, glutathione peroxidase-like protein, tryparedoxin 1 (TXN1), iron superoxide dismutase |
| Flagellar proteins | PFR2, PFR1, KHAP1, flagellum targeting protein kharon1 |
| Proteases | Aminopeptidase, carboxypeptidase CP1, calpain-like cysteine peptidase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adán-Jiménez, J.; Sánchez-Salvador, A.; Morato, E.; Solana, J.C.; Aguado, B.; Requena, J.M. A Proteogenomic Approach to Unravel New Proteins Encoded in the Leishmania donovani (HU3) Genome. Genes 2024, 15, 775. https://doi.org/10.3390/genes15060775
Adán-Jiménez J, Sánchez-Salvador A, Morato E, Solana JC, Aguado B, Requena JM. A Proteogenomic Approach to Unravel New Proteins Encoded in the Leishmania donovani (HU3) Genome. Genes. 2024; 15(6):775. https://doi.org/10.3390/genes15060775
Chicago/Turabian StyleAdán-Jiménez, Javier, Alejandro Sánchez-Salvador, Esperanza Morato, Jose Carlos Solana, Begoña Aguado, and Jose M. Requena. 2024. "A Proteogenomic Approach to Unravel New Proteins Encoded in the Leishmania donovani (HU3) Genome" Genes 15, no. 6: 775. https://doi.org/10.3390/genes15060775