
Transcriptomic Insights into the Developmental Dynamics of Eimeria acervulina: A Comparative Study of a Precocious Line and the Wild Type
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Parasites
2.2. Preparation of Samples for RNA-Seq
2.3. RNA Sequencing and Bioinformatic Analyses
3. Results
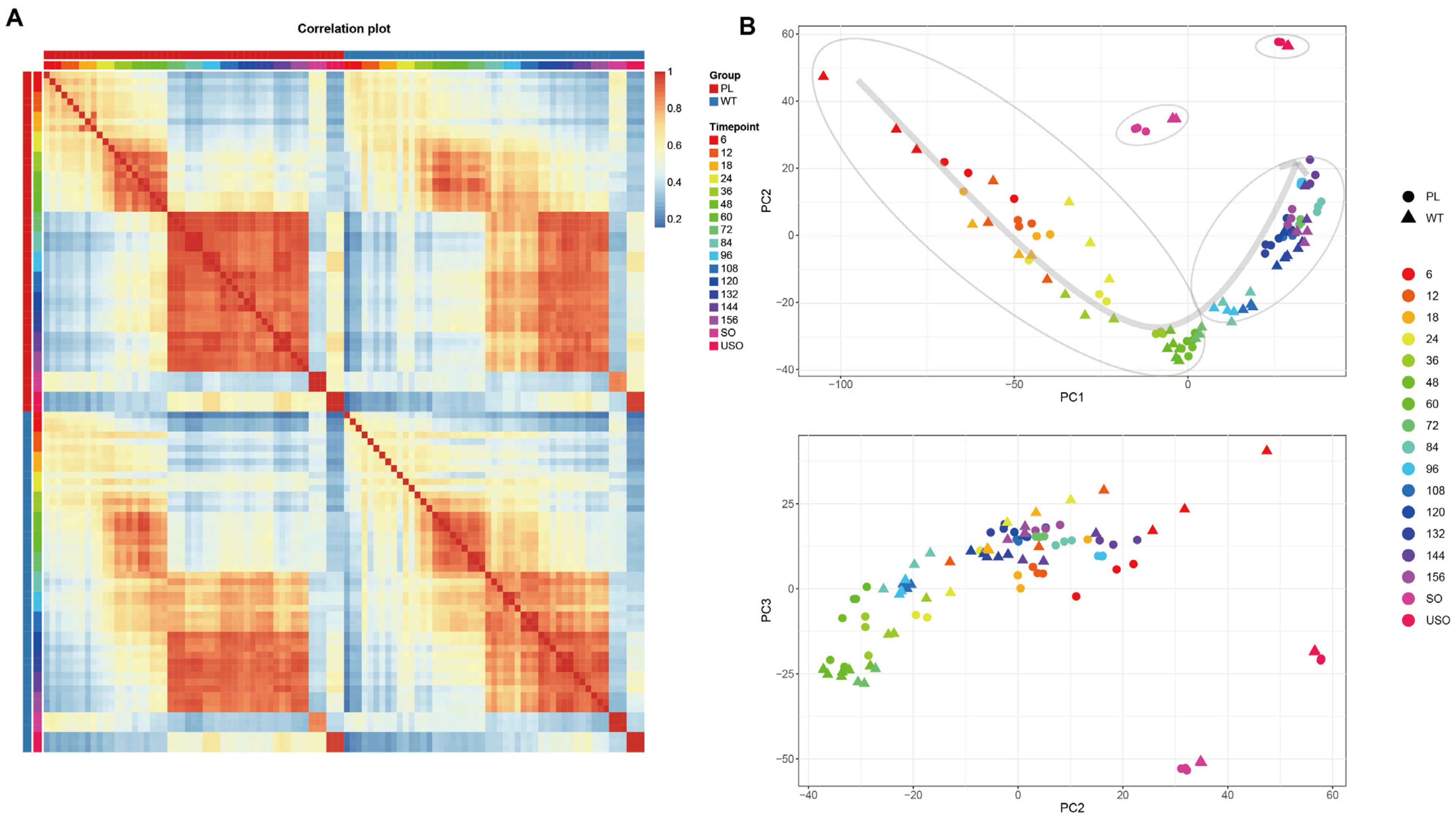
3.1. Similar but Accelerated Gene Expression Dynamics in the Precocious Line of E. acervulina
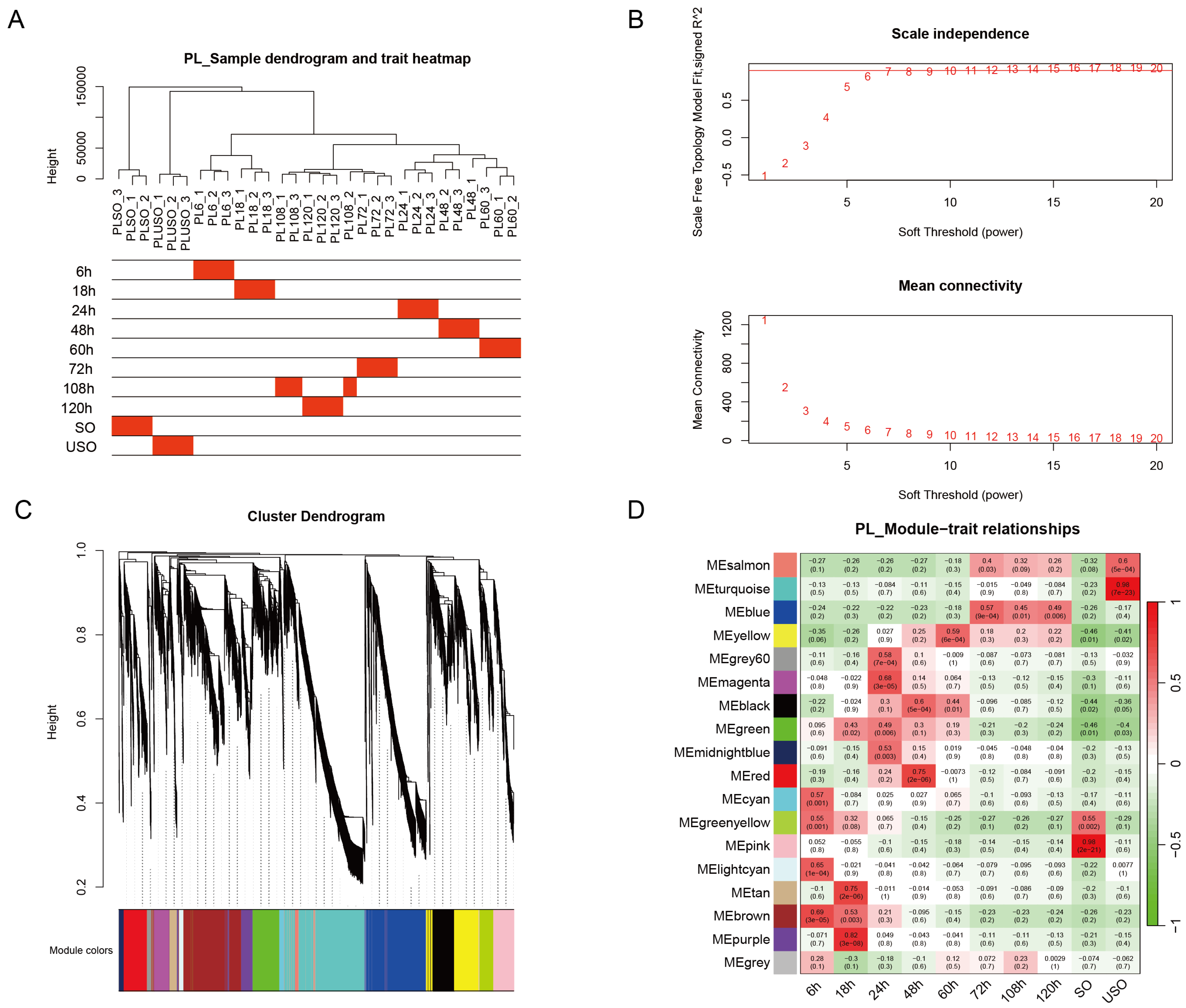
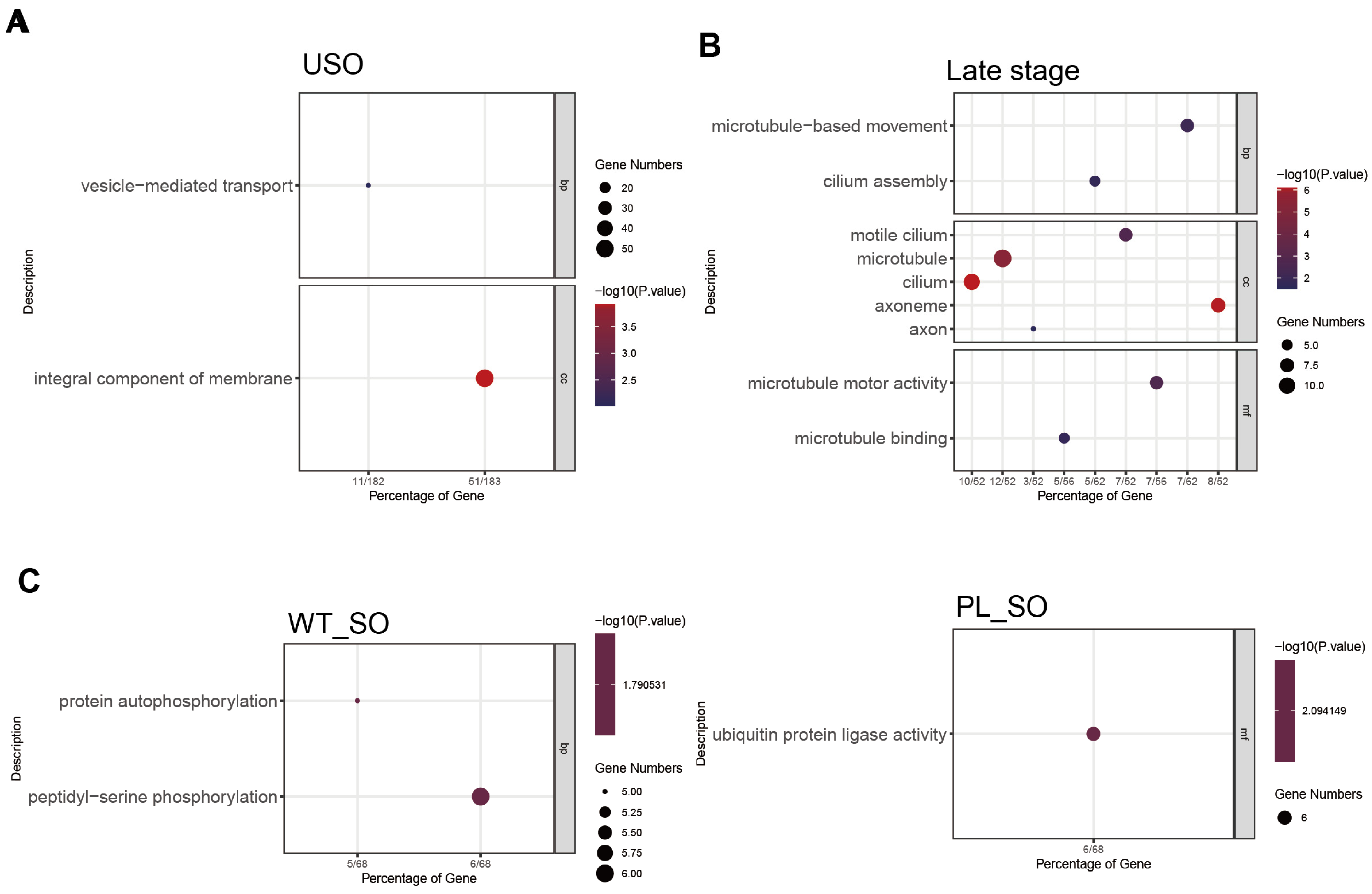
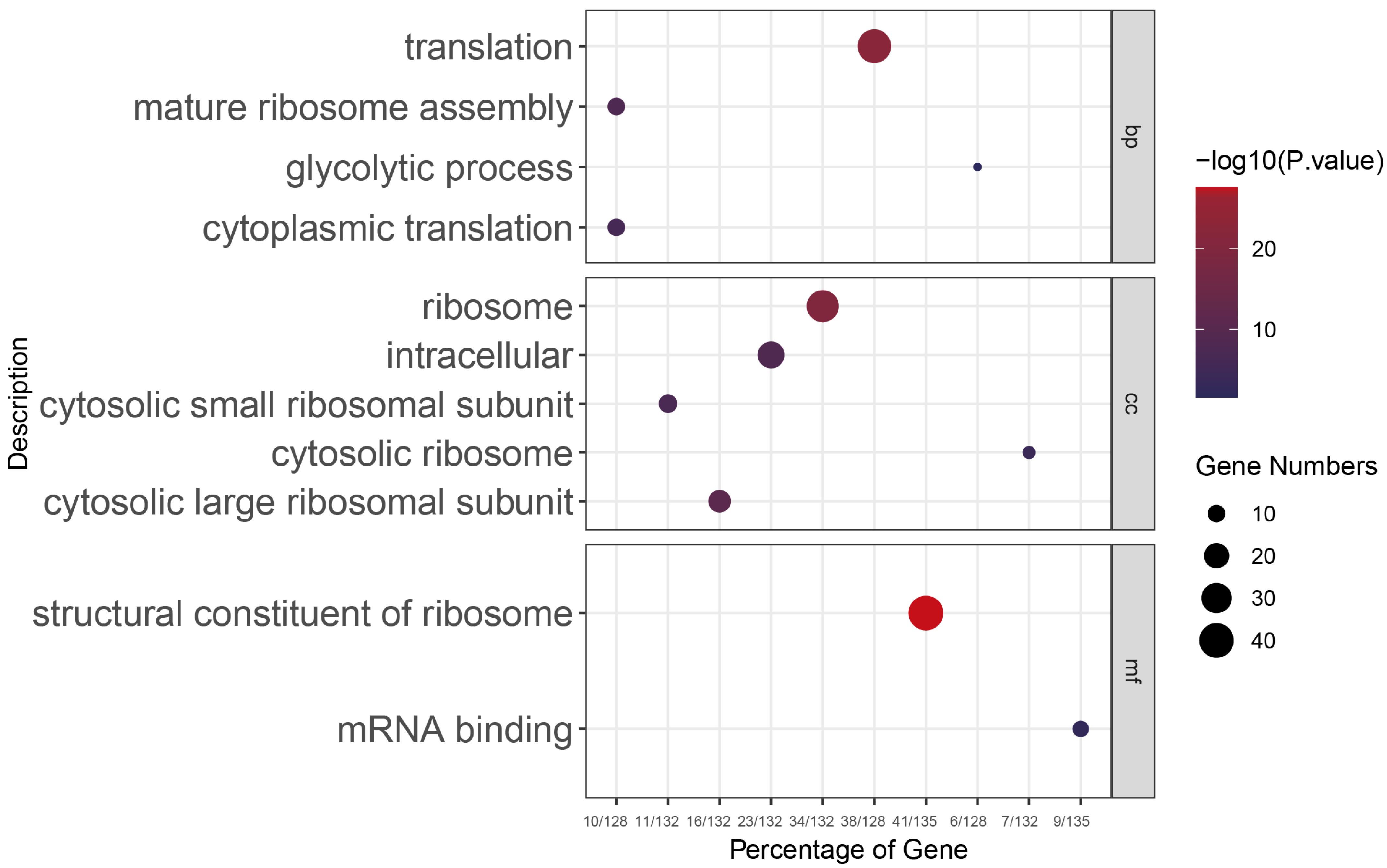
3.2. Key Regulatory Pathways in E. acervulina Developmental Stages Identified through Comparative Gene Expression Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blake, D.P.; Vrba, V.; Xia, D.; Jatau, I.D.; Jatau, I.D.; Spiro, S.; Nolan, M.J.; Underwood, G.; Tomley, F.M. Genetic and biological characterization of three cryptic Eimeria operational taxonomic units that infect chickens (Gallus gallus domesticus). Int. J. Parasitol. 2021, 51, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Sun, P.; Hu, D.; Tang, X.; Zhang, S.; Shi, F.; Yan, X.; Yan, W.; Shi, T.; Wang, S.; et al. Advancements in understanding chicken coccidiosis: From Eimeria biology to innovative control strategies. One Health Adv. 2024, in press.
- Blake, D.P.; Worthing, K.; Jenkins, M.C. Exploring Eimeria Genomes to Understand Population Biology: Recent Progress and Future Opportunities. Genes 2020, 11, 1103. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.; Knox, J.; Dehaeck, B.; Huntington, B.; Rathinam, T.; Ravipati, V.; Ayoade, S.; Gilbert, W.; Adebambo, A.O.; Jatau, I.D.; et al. Re-Calculating the cost of coccidiosis in chickens. Vet. Res. 2020, 51, 115. [Google Scholar] [CrossRef]
- Saeed, Z.; Alkheraije, K.A. Botanicals: A promising approach for controlling cecal coccidiosis in poultry. Front. Vet. Sci. 2023, 10, 1157633. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Liu, H.; Li, C.; Fang, S.; Cui, P.; Liao, Q.; Zhang, S.; Wang, S.; Duan, C.; Yu, F.; et al. Selection and characterization of a precocious line of Eimeria media. Parasitol. Res. 2019, 118, 3033–3041. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.J.; Pittilo, R.M.; Joyner, L.P.; Norton, C.C. Scanning and transmission electron microscopy of Eimeria maxima microgametogenesis. Parasitology 1981, 82, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Novilla, M.N.; Jeffers, T.K.; Griffing, W.J.; White, S.L. A redescription of the life cycle of Eimeria mitis Tyzzer, 1929. J. Protozool. 1987, 34, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Kawazoe, U.; Bordin, E.L.; de Lima, C.A.; Dias, L.A. Characterisation and histopathological observations of a selected Brazilian precocious line of Eimeria acervulina. Vet. Parasitol. 2005, 131, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Vetterling, J.M.; Doran, D.J. Schizogony and gametogony in the life cycle of the poultry coccidium, Eimeria acervulina Tyzzer, 1929. J. Parasitol. 1966, 52, 1150–1157. [Google Scholar] [CrossRef]
- Behnke, M.S.; Zhang, T.P.; Dubey, J.P.; Sibley, L.D. Toxoplasma gondii merozoite gene expression analysis with comparison to the life cycle discloses a unique expression state during enteric development. BMC Genom. 2014, 15, 350. [Google Scholar]
- Fritz, H.M.; Buchholz, K.R.; Chen, X.; Durbin-Johnson, B.; Rocke, D.M.; Conrad, P.A.; Boothroyd, J.C. Transcriptomic analysis of Toxoplasma development reveals many novel functions and structures specific to sporozoites and oocysts. PLoS ONE 2012, 7, e29998. [Google Scholar] [CrossRef] [PubMed]
- Silvestrini, F.; Bozdech, Z.; Lanfrancotti, A.; Giulio, E.D.; Bultrini, E.; Picci, L.; deRisi, J.L.; Pizzi, E.; Alano, P. Genome-wide identification of genes upregulated at the onset of gametocyte genesis in Plasmodium falciparum. Mol. Biochem. Parasitol. 2005, 143, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Ferguson, D.J.; Blackman, M.J.; Soldati-Favre, D. Intramembrane cleavage of AMA1 triggers Toxoplasma to switch from an invasive to a replicative mode. Science 2011, 331, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Modrzynska, K.; Pfander, C.; Chappell, L.; Yu, L.; Suarez, C.; Dundas, K.; Gomes, A.R.; Goulding, D.; Rayner, J.C.; Choudhary, J.; et al. A Knockout Screen of ApiAP2 Genes Reveals Networks of Interacting Transcriptional Regulators Controlling the Plasmodium Life Cycle. Cell Host Microbe 2017, 21, 11–22. [Google Scholar] [CrossRef]
- Iwanaga, S.; Kaneko, I.; Kato, T.; Yuda, M. Identification of an AP2-family protein that is critical for malaria liver stage development. PLoS ONE 2012, 7, e47557. [Google Scholar] [CrossRef] [PubMed]
- Amiruddin, N.; Lee, X.W.; Blake, D.P.; Suzuki, Y.; Tay, Y.L.; Lim, L.S.; Tomley, F.M.; Watanabe, J.; Sugimoto, C.; Wan, K.-L. Characterisation of full-length cDNA sequences provides insights into the Eimeria tenella transcriptome. BMC Genom. 2012, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, M.; Hatta, T.; Miyoshi, T.; Anisuzzaman, S.K.; Shimura, K.; Isobe, T.; Kita, K.; Tsuji, N. High-throughput RNA sequencing profiles and transcriptional evidence of aerobic respiratory enzymes in sporulating oocysts and sporozoites of Eimeria tenella. Infect. Genet. Evol. 2013, 18, 269–276. [Google Scholar] [CrossRef]
- Novaes, J.; Rangel, L.T.; Ferro, M.; Abe, R.Y.; Manha, A.P.; de Mello, J.C.; Varuzza, L.; Durham, A.M.; Madeira, A.M.B.; Gruber, A. A comparative transcriptome analysis reveals expression profiles conserved across three Eimeria spp. of domestic fowl and associated with multiple developmental stages. Int. J. Parasitol. 2012, 42, 39–48. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Blighe, K.; Lun, A. PCAtools: Everything Principal Components Analysis. 2019. Available online: https://github.com/kevinblighe/PCAtools (accessed on 19 June 2024).
- Li, C.; Zhao, Q.; Zhu, S.; Wang, Q.; Wang, H.; Yu, S.; Yu, Y.; Liang, S.; Zhao, H.; Huang, B.; et al. Eimeria tenella Eimeria-specific protein that interacts with apical membrane antigen 1 (EtAMA1) is involved in host cell invasion. Parasites Vectors 2020, 13, 373. [Google Scholar] [CrossRef]
- Zhao, N.; Ming, S.; Sun, L.; Wang, B.; Li, H.; Zhang, X.; Zhao, X. Identification and Characterization of Eimeria tenella Microneme Protein (EtMIC8). Microbiol. Spectr. 2021, 9, e0022821. [Google Scholar] [CrossRef] [PubMed]
- Augustine, P.C. Cell: Sporozoite interactions and invasion by apicomplexan parasites of the genus Eimeria. Int. J. Parasitol. 2001, 31, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Suding, Z.; Wang, L.; Liu, D.; Su, S.; Xu, J.; Hu, J.; Tao, J. Full-length transcriptome analysis and identification of transcript structures in Eimeria necatrix from different developmental stages by single-molecule real-time sequencing. Parasites Vectors 2021, 14, 502. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Boothroyd, J.C. Stage-specific expression of surface antigens by Toxoplasma gondii as a mechanism to facilitate parasite persistence. J. Immunol. 2005, 174, 8038–8048. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Y.; Liang, Y.; Wang, S.; Xie, Q.; Nan, X.; Li, P.; Hong, G.; Liu, Q.; Li, X. Molecular characterization and protective immunity of rhoptry protein 35 (ROP35) of Toxoplasma gondii as a DNA vaccine. Vet. Parasitol. 2018, 260, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Hou, Z.; Liu, D.; Jia, C.; Wang, L.; Xu, J.; Tao, J. Comparative transcriptome analysis of second- and third-generation merozoites of Eimeria necatrix. Parasites Vectors 2017, 10, 388. [Google Scholar] [CrossRef]
- Su, S.; Hou, Z.; Liu, D.; Jia, C.; Wang, L.; Xu, J.; Tao, J. Comparative transcriptome analysis of Eimeria necatrix third-generation merozoites and gametocytes reveals genes involved in sexual differentiation and gametocyte development. Vet. Parasitol. 2018, 252, 35–46. [Google Scholar] [CrossRef]
- Walker, R.A.; Sharman, P.A.; Miller, C.M.; Lippuner, C.; Okoniewski, M.; Eichenberger, R.M.; Ramakrishnan, C.; Brossier, F.; Deplazes, P.; Hehl, A.B.; et al. RNA Seq analysis of the Eimeria tenella gametocyte transcriptome reveals clues about the molecular basis for sexual reproduction and oocyst biogenesis. BMC Genom. 2015, 16, 94. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, N.; Li, X.; Liu, J.; Chen, L.; Zhang, S.; Liu, X.; Tang, X.; Suo, X.; Zhang, Y. Transcriptomic Insights into the Developmental Dynamics of Eimeria acervulina: A Comparative Study of a Precocious Line and the Wild Type. Genes 2024, 15, 831. https://doi.org/10.3390/genes15070831
Zhang N, Li X, Liu J, Chen L, Zhang S, Liu X, Tang X, Suo X, Zhang Y. Transcriptomic Insights into the Developmental Dynamics of Eimeria acervulina: A Comparative Study of a Precocious Line and the Wild Type. Genes. 2024; 15(7):831. https://doi.org/10.3390/genes15070831
Chicago/Turabian StyleZhang, Ning, Xiaojin Li, Jie Liu, Linlin Chen, Sixin Zhang, Xianyong Liu, Xinming Tang, Xun Suo, and Yuanyuan Zhang. 2024. "Transcriptomic Insights into the Developmental Dynamics of Eimeria acervulina: A Comparative Study of a Precocious Line and the Wild Type" Genes 15, no. 7: 831. https://doi.org/10.3390/genes15070831