
Sequencing and Description of the Mitochondrial Genome of Orthopodomyia fascipes (Diptera: Culicidae)

 , ,
, ,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Total DNA Extraction
2.2. Genomic Library Preparation and Sequencing
2.3. Data Processing and mtDNA Characterization
2.4. Phylogenetic Analysis
3. Results
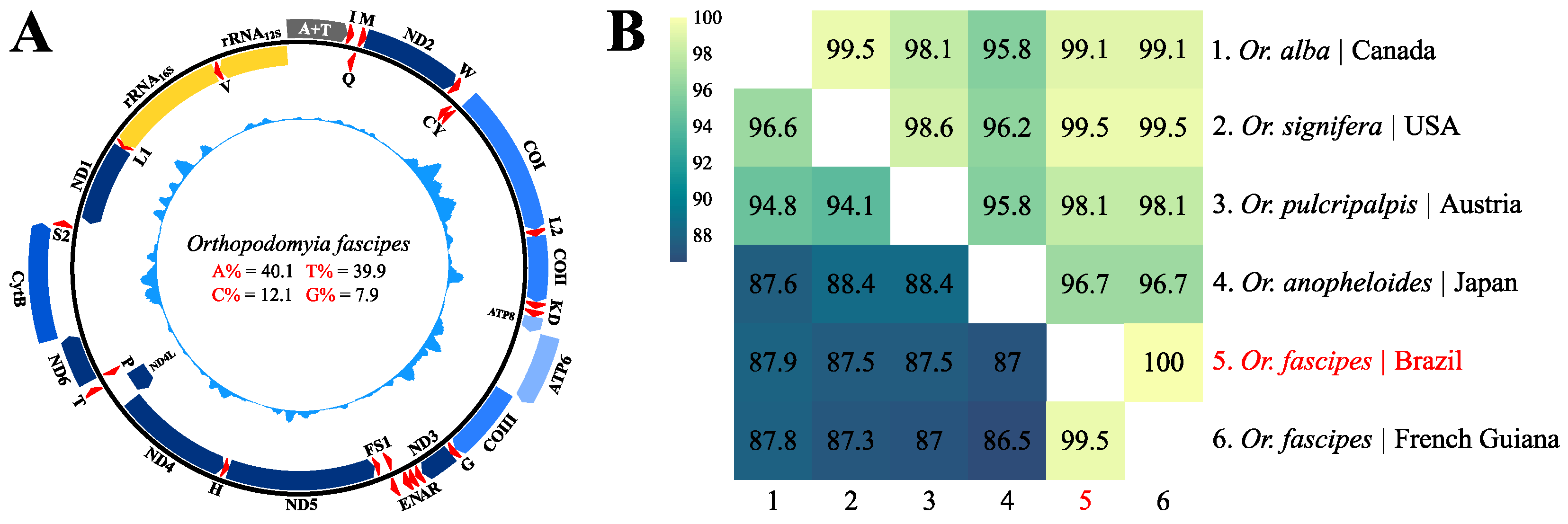
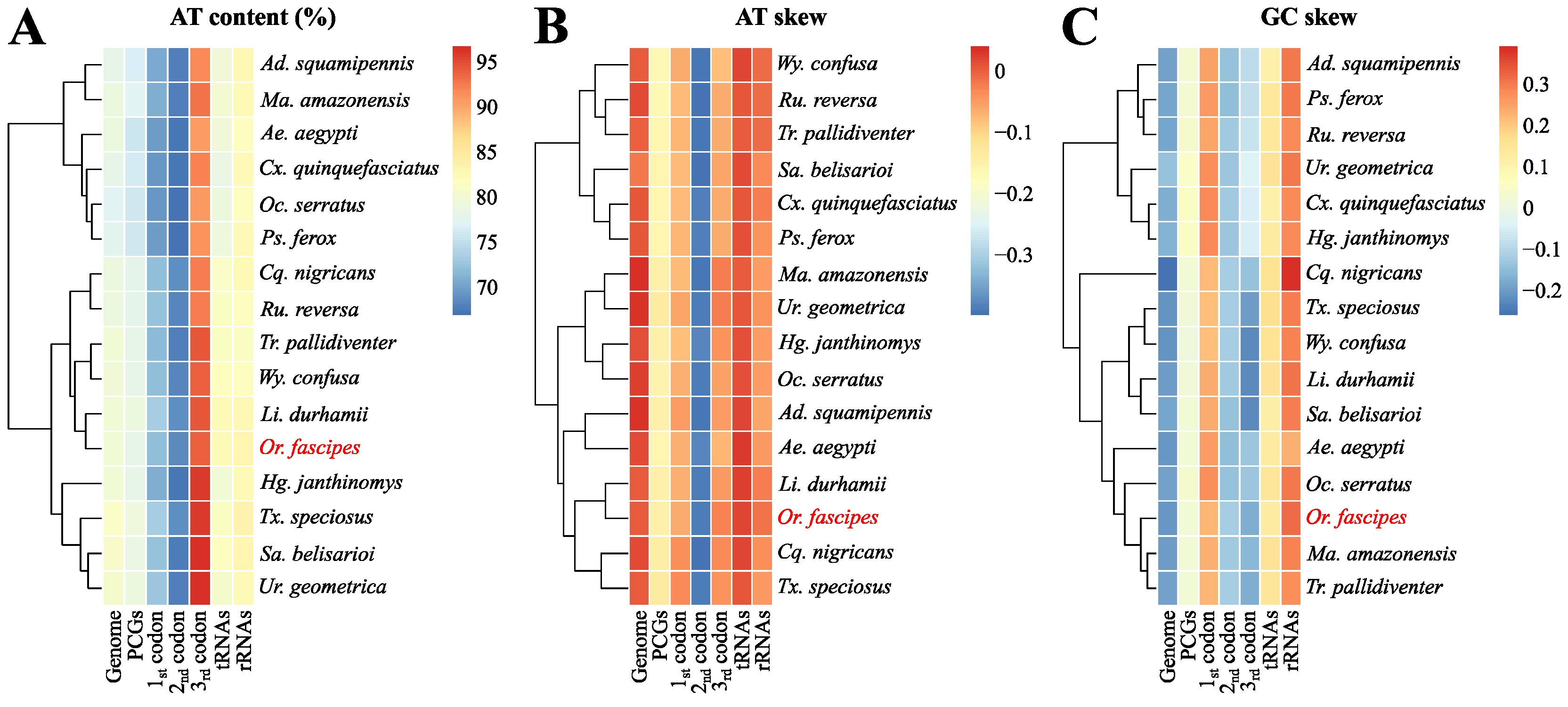
3.1. Assembly, Genomic Organization and General Composition of the Sequence Obtained
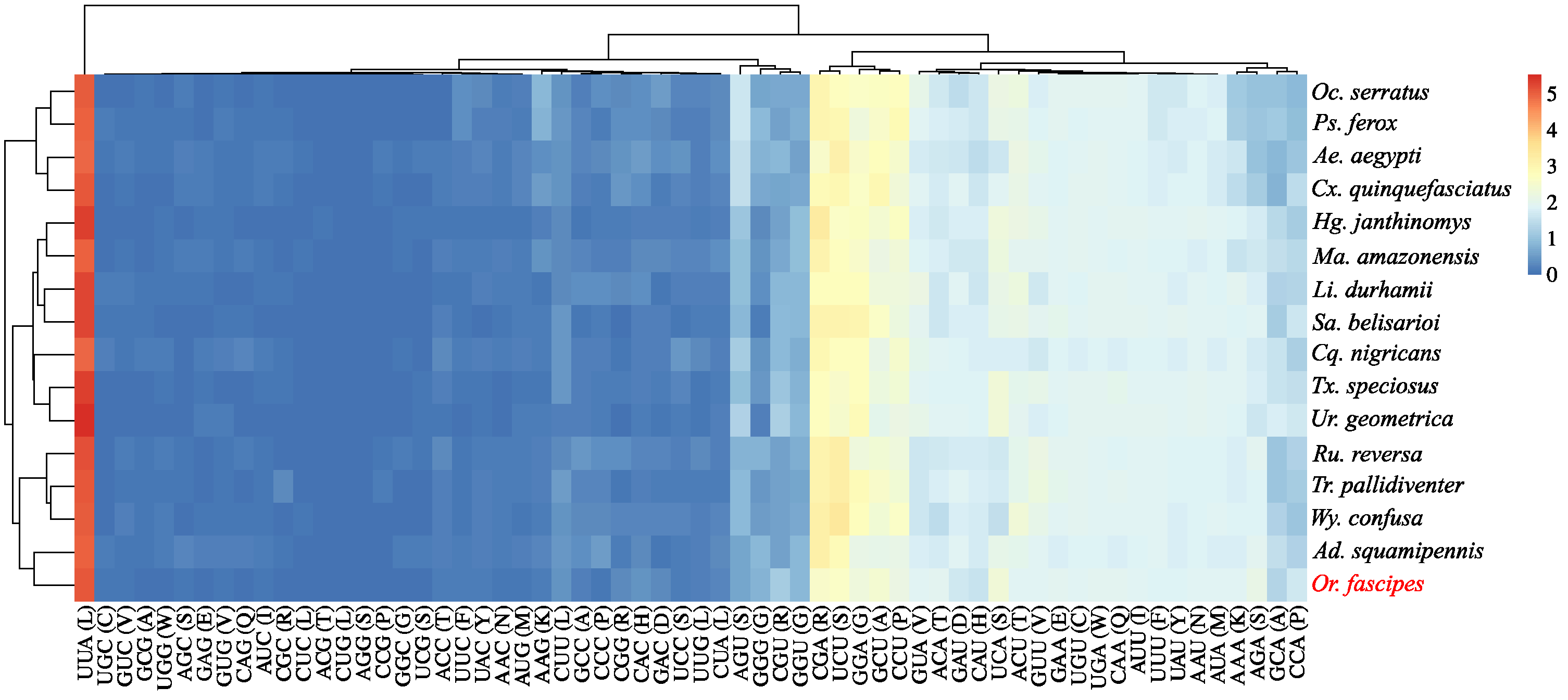
3.2. Description of Protein-Coding Genes (PCGs)
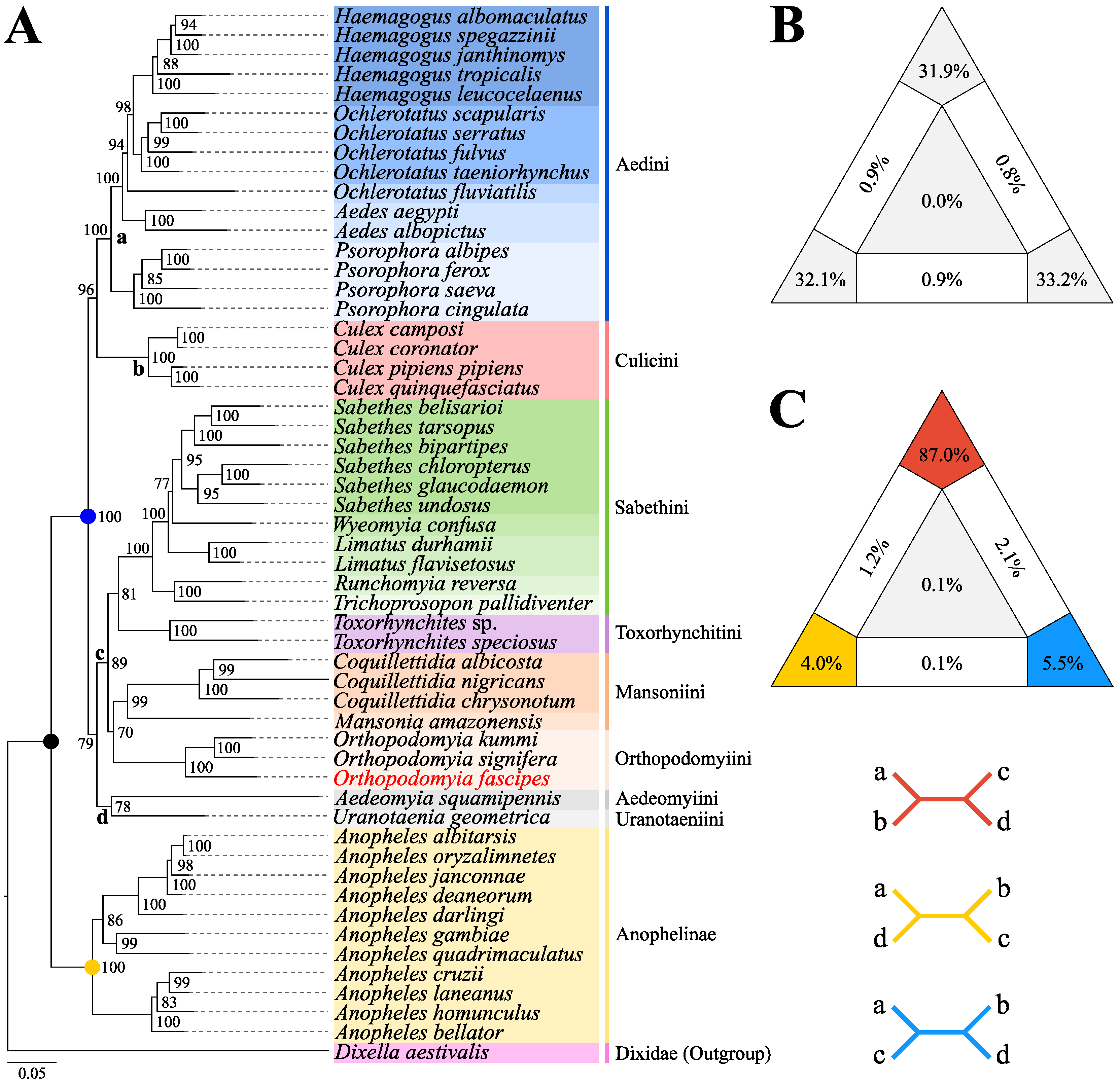
3.3. Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amorim, D.d.S.; Yeates, D. Pesky Gnats: Ridding Dipteran Classification of the Nematocera. Stud. Dipterol. 2006, 13, 1–7. [Google Scholar]
- Kutty, S.N.; Wong, W.H.; Meusemann, K.; Meier, R.; Cranston, P.S. A Phylogenomic Analysis of Culicomorpha (Diptera) Resolves the Relationships among the Eight Constituent Families. Syst. Entomol. 2018, 43, 434–446. [Google Scholar] [CrossRef]
- Harbach, R.E. The Culicidae (Diptera): A Review of Taxonomy, Classification and Phylogeny. Zootaxa 2007, 638, 591–638. [Google Scholar] [CrossRef]
- Harbach, R.E. Mosquito Taxonomic Inventory. Available online: https://mosquito-taxonomic-inventory.myspecies.info/ (accessed on 11 April 2024).
- Forattini, O.P. Culicidologia Médica: Identificação, Biologia, Epidemiologia; Edusp: São Paulo, Brazil, 2002; Volume 2. [Google Scholar]
- Reidenbach, K.R.; Cook, S.; Bertone, M.A.; Harbach, R.E.; Wiegmann, B.M.; Besansky, N.J. Phylogenetic Analysis and Temporal Diversification of Mosquitoes (Diptera: Culicidae) Based on Nuclear Genes and Morphology. BMC Evol. Biol. 2009, 9, 298. [Google Scholar] [CrossRef] [PubMed]
- Soghigian, J.; Andreadis, T.G.; Livdahl, T.P. From Ground Pools to Treeholes: Convergent Evolution of Habitat and Phenotype in Aedes Mosquitoes. BMC Evol. Biol. 2017, 17, 262. [Google Scholar] [CrossRef] [PubMed]
- Lobo, F.P.; Mota, B.E.F.; Pena, S.D.J.; Azevedo, V.; Macedo, A.M.; Tauch, A.; Machado, C.R.; Franco, G.R. Virus-Host Coevolution: Common Patterns of Nucleotide Motif Usage in Flaviviridae and Their Hosts. PLoS ONE 2009, 4, e6282. [Google Scholar] [CrossRef] [PubMed]
- Severson, D.W.; Behura, S.K. Genome Investigations of Vector Competence in Aedes aegypti to Inform Novel Arbovirus Disease Control Approaches. Insects 2016, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Behura, S.K. Molecular Marker Systems in Insects: Current Trends and Future Avenues. Mol. Ecol. 2006, 15, 3087–3113. [Google Scholar] [CrossRef]
- Ditter, R.E.; Campos, M.; Pinto, J.; Cornel, A.J.; Rompão, H.; Lanzaro, G.C. Mitogenome Analyses Reveal Limited Introduction of Anopheles coluzzii Into the Central African Islands of São Tomé and Príncipe. Front. Trop. Dis. 2022, 3, 855272. [Google Scholar] [CrossRef]
- Patz, J.A.; Graczyk, T.K.; Geller, N.; Vittor, A.Y. Environmental Changes & Parasitic Diseases. Int. J. Parasitol. 2000, 30, 1395–1405. [Google Scholar]
- Vasconcelos, P.F.; Travassos da Rosa, A.P.; Rodrigues, S.G.; Travassos da Rosa, E.S.; Dégallier, N.; Travassos da Rosa, J.F. Inadequate Management of Natural Ecosystem in the Brazilian Amazon Region Results in the Emergence and Reemergence of Arboviruses. Cad. Saúde Pública/Ministério Saúde Fundação Oswaldo Cruz Esc. Nac. Saúde Pública 2001, 17, 155–164. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global Trends in Emerging Infectious Diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, D.; Piche-Ovares, M.; Romero-Vega, L.M.; Wagman, J.; Troyo, A. The Impact of Deforestation, Urbanization, and Changing Land Use Patterns on the Ecology of Mosquito and Tick-Borne Diseases in Central America. Insects 2022, 13, 20. [Google Scholar] [CrossRef]
- Zavortink, T.J. Mosquito Studies (Diptera, Culicidae) VIII. A Prodrome of the Genus Orthopodomyia. Contrib. Am. Entomol. Inst. 1968, 3, 221. [Google Scholar]
- Lane, J. Neotropical Culicidae; University of São Paulo: São Paulo, Brazil, 1953. [Google Scholar]
- Yeo, H.; Tan, C.H.; Chong, C.S.; Lam-Phua, S.G. Identification Key to the Genera of Adult Female Mosquitoes (Diptera: Culicidae) of Singapore. Zootaxa 2019, 4624, 407–423. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.M.; Novak, R.J.; Lampman, R.L.; Vodkin, M.H. Notes on the Biology of Orthopodomyia in Illinois. J. Am. Mosq. Control Assoc. 1995, 11, 375–376. [Google Scholar] [PubMed]
- Becker, N.; Petric, D.; Zgomba, M.; Boase, C.; Madon, M.; Dahl, C.; Kaiser, A. Mosquitoes and Their Control, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Chamberlain, R.W.; Sikes, R.K.; Nelson, D.B.; Sudia, W.D. Studies on the North American Arthropod-Borne Encephalitides. VI. Quantitative Determinations of Virus-Vector Relationships. Am. J. Trop. Med. Hyg. 1954, 60, 278–285. [Google Scholar] [CrossRef]
- Vargas, L. Los Mosquitos de Sonora En Relacion Con El Problem de Encephalitis. Rev. Mex. Med. 1960, 40, 338–345. [Google Scholar]
- Centers for Disease Control and Prevention (CDC). West Nile Virus. Available online: https://www.cdc.gov/westnile/vectorcontrol/index.html (accessed on 17 April 2024).
- Edwards, F.W. Genera Insectorum. Diptera, Fam. Culicidae. Fascicle 194; Desmet-Verteneuil: Brussels, Belgium, 1932. [Google Scholar]
- Harbach, R.E.; Kitching, I.J. Phylogeny and Classification of the Culicidae (Diptera). Syst. Entomol. 1998, 23, 327–370. [Google Scholar] [CrossRef]
- Yan, G.; Chadee, D.D.; Severson, D.W. Evidence for Genetic Hitchhiking Effect Associated with Insecticide Resistance in Aedes aegypti. Genetics 1998, 148, 793–800. [Google Scholar] [CrossRef]
- Barzon, L.; Lavezzo, E.; Militello, V.; Toppo, S.; Palù, G. Applications of Next-Generation Sequencing Technologies to Diagnostic Virology. Int. J. Mol. Sci. 2011, 12, 7861–7884. [Google Scholar] [CrossRef]
- Tabachnick, W.J. Nature, Nurture and Evolution of Intra-Species Variation in Mosquito Arbovirus Transmission Competence. Int. J. Environ. Res. Public Health 2013, 10, 249–277. [Google Scholar] [CrossRef]
- Brown, W.M.; George, M.; Wilson, A.C. Rapid Evolution of Animal Mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef]
- Avise, J.C. Phylogeography: Retrospect and Prospect. J. Biogeogr. 2009, 36, 3–15. [Google Scholar] [CrossRef]
- Boore, J.L. Animal Mitochondrial Genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Mandal, S.D.; Chhakchhuak, L.; Gurusubramanian, G.; Kumar, N.S. Mitochondrial Markers for Identification and Phylogenetic Studies in Insects—A Review. DNA Barcodes 2014, 2, 1–9. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological Identifications through DNA Barcodes. Proc. R. Soc. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Hao, Y.J.; Zou, Y.L.; Ding, Y.R.; Xu, W.Y.; Yan, Z.T.; Li, X.D.; Fu, W.B.; Li, T.J.; Chen, B. Complete Mitochondrial Genomes of Anopheles stephensi and An. dirus and Comparative Evolutionary Mitochondriomics of 50 Mosquitoes. Sci. Rep. 2017, 7, 7666. [Google Scholar] [CrossRef]
- Demari-Silva, B.; Foster, P.G.; de Oliveira, T.M.P.; Bergo, E.S.; Sanabani, S.S.; Pessôa, R.; Sallum, M.A.M. Mitochondrial Genomes and Comparative Analyses of Culex camposi, Culex coronator, Culex usquatus and Culex usquatissimus (Diptera: Culicidae), Members of the Coronator Group. BMC Genomics 2015, 16, 831. [Google Scholar] [CrossRef]
- Lorenz, C.; Alves, J.M.P.; Foster, P.G.; Sallum, M.A.M.; Suesdek, L. First Record of Translocation in Culicidae (Diptera) Mitogenomes: Evidence from the Tribe Sabethini. BMC Genomics 2019, 20, 721. [Google Scholar] [CrossRef]
- Silva, F.S.; Cruz, A.C.R.; Medeiros, D.B.d.A.; Silva, S.P.; Nunes, M.R.T.; Martins, L.C.; Chiang, J.O.; Lemos, P.d.S.; Cunha, G.M.; Araujo, R.F.; et al. Mitochondrial Genome Sequencing and Phylogeny of Haemagogus albomaculatus, Haemagogus leucocelaenus, Haemagogus spegazzinii, and Haemagogus tropicalis (Diptera: Culicidae). Sci. Rep. 2020, 10, 16948. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, B.L.S.; Silva, F.S.; Nunes-Neto, J.P.; Medeiros, D.B.d.A.; Cruz, A.C.R.; Silva, S.P.; Silva, L.H.d.S.; Monteiro, H.A.d.O.; Dias, D.D.; Vieira, D.B.R.; et al. First Description of the Mitogenome and Phylogeny of Culicinae Species from the Amazon Region. Genes 2021, 12, 1983. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Alves, J.M.P.; Foster, P.G.; Suesdek, L.; Sallum, M.A.M. Phylogeny and Temporal Diversification of Mosquitoes (Diptera: Culicidae) with an Emphasis on the Neotropical Fauna. Syst. Entomol. 2021, 46, 798–811. [Google Scholar] [CrossRef]
- Silva, L.H.d.S.; Silva, F.S.; Medeiros, D.B.d.A.; Cruz, A.C.R.; Silva, S.P.; Aragão, A.d.O.; Dias, D.D.; Nascimento, B.L.S.; Rosa, J.W., Jr.; Vieira, D.B.R.; et al. Description of the Mitogenome and Phylogeny of Aedes spp. (Diptera: Culicidae) from the Amazon Region. Acta Trop. 2022, 232, 106500. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.H.; He, S.L.; Fu, W.B.; Yan, Z.T.; Hu, Y.J.; Yuan, H.; Wang, M.B.; Chen, B. Mitogenome-Based Phylogeny of Mosquitoes (Diptera: Culicidae). Insect Sci. 2024, 31, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.S.; Nascimento, B.L.S.; Cruz, A.C.R.; Silva, S.P.; Aragão, A.d.O.; Dias, D.D.; Silva, L.H.d.S.; Reis, L.A.M.; Rosa, J.W.; Vieira, D.B.R.; et al. Sequencing and Description of the Complete Mitochondrial Genome of Limatus durhamii (Diptera: Culicidae). Acta Trop. 2023, 239, 106805. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.A.d.; Cruz, A.C.R.; Silva, F.S.; Silva, S.P.; Nunes-Neto, J.P.; Barros, M.C.; Fraga, E.d.C.; Sampaio, I. Sequencing and Analysis of the Mitochondrial Genome of Aedes aegypti (Diptera: Culicidae) from the Brazilian Amazon Region. Insects 2023, 14, 938. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information (NCBI). Orthopodomyia. Available online: https://www.ncbi.nlm.nih.gov/datasets/taxonomy/139053/ (accessed on 11 April 2024).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive Metagenomic Visualization in a Web Browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de Novo Metazoan Mitochondrial Genome Annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and Comparing Circular Genomes Using the CGView Family of Tools. Brief. Bioinform. 2018, 20, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of Nucleotide Composition at Fourfold Degenerate Sites of Animal Mitochondrial Genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 1999. [Google Scholar]
- Yang, Z. User Guide PAML: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 4, 1–70. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 6 January 2024).
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize Implements and Enhances Circular Visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2 Elegant Graphics for Data Analysis, 2nd ed.; Gentleman, R., Hornik, K., Parmigiani, G., Eds.; Springer: Houston, TX, USA, 2016. [Google Scholar]
- Kolde, R. Package “Pheatmap”: Pretty Heatmaps. R Package Version 1.0.12. Available online: https://cran.r-project.org/web/packages/pheatmap/ (accessed on 30 March 2024).
- Wickham, H. Reshaping Data with the Reshape Package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A Fast and Lightweight Alignment Viewer and Editor for Large Datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An Index of Substitution Saturation and Its Application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE7: New and Improved Tools for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R Language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Strimmer, K.; Von Haeseler, A. Likelihood-Mapping: A Simple Method to Visualize Phylogenetic Content of a Sequence Alignment. Proc. Natl. Acad. Sci. USA 1997, 94, 6815–6819. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v.1.4.4. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 6 January 2024).
- Cameron, S.L. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. TRNA Punctuation Model of RNA Processing in Human Mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wan, D.G.; Murphy, R.W.; Liang, M.; Zhang, X.S.; Huang, D.W. Comparison of Base Composition and Codon Usage in Insect Mitochondrial Genomes. Genes Genomics 2009, 31, 65–71. [Google Scholar] [CrossRef]
- Wang, H.; Meng, T.; Wei, W. Analysis of Synonymous Codon Usage Bias in Helicase Gene from Autographa Californica Multiple Nucleopolyhedrovirus. Genes Genomics 2018, 40, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The “effective Number of Codons” Used in a Gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Sueoka, N. Directional Mutation Pressure and Neutral Molecular Evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef]
- Zhangtl, Z.; Yu, J. Evaluation of Six Methods for Estimating Synonymous and Non-Synonymous Substitution Rates Comparative Results. Genomics Proteomics Bioinform. 2006, 4, 173–181. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal Mitochondrial DNA: Structure and Evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-Y.; Deng, Y.-P.; Zhang, Y.; Wu, Y.; Fu, Y.-T.; Liu, G.-H.; Liu, J.-H. Characterization of the Complete Mitochondrial Genome of Culex vishnui (Diptera: Culicidae), One of the Major Vectors of Japanese Encephalitis Virus. Parasitol. Res. 2023, 122, 1403–1414. [Google Scholar] [CrossRef]
- Soghigian, J.; Sither, C.; Justi, S.A.; Morinaga, G.; Cassel, B.K.; Vitek, C.J.; Livdahl, T.; Xia, S.; Gloria-Soria, A.; Powell, J.R.; et al. Phylogenomics Reveals the History of Host Use in Mosquitoes. Nat. Commun. 2023, 14, 6252. [Google Scholar] [CrossRef]
- Silva, A.F.; Machado, L.C.; de Paula, M.B.; Vieira, C.J.d.S.P.; Bronzoni, R.V.d.M.; Santos, M.A.V.d.M.; Wallau, G.L. Culicidae Evolutionary History Focusing on the Culicinae Subfamily Based on Mitochondrial Phylogenomics. Sci. Rep. 2020, 10, 18823. [Google Scholar] [CrossRef]
- Munstermann, L.E.; Marchi, A.; Sabatini, A.; Coluzzi, M. Polytene Chromosomes of Orthopodomyia pulcripalpis (Diptera, Culicidae). Parassitologia 1985, 27, 267–277. [Google Scholar] [PubMed]
- Coquillett, D.W. New Culicidae from the West Indies and Central America. Proc. Entomol. Soc. Wash. 1906, 7, 182–186. [Google Scholar]
- Kjer, K.M.; Simon, C.; Yavorskaya, M.; Beutel, R.G. Progress, Pitfalls and Parallel Universes: A History of Insect Phylogenetics. J. R. Soc. Interface 2016, 13, 20160363. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, F.S.d.; Nascimento, B.L.S.d.; Cruz, A.C.R.; Silva, S.P.d.; Aragão, C.F.; Dias, D.D.; Silva, L.H.d.S.e.; Reis, L.A.M.; Reis, H.C.F.; Chagas, L.L.d.; et al. Sequencing and Description of the Mitochondrial Genome of Orthopodomyia fascipes (Diptera: Culicidae). Genes 2024, 15, 874. https://doi.org/10.3390/genes15070874
Silva FSd, Nascimento BLSd, Cruz ACR, Silva SPd, Aragão CF, Dias DD, Silva LHdSe, Reis LAM, Reis HCF, Chagas LLd, et al. Sequencing and Description of the Mitochondrial Genome of Orthopodomyia fascipes (Diptera: Culicidae). Genes. 2024; 15(7):874. https://doi.org/10.3390/genes15070874
Chicago/Turabian StyleSilva, Fábio Silva da, Bruna Laís Sena do Nascimento, Ana Cecília Ribeiro Cruz, Sandro Patroca da Silva, Carine Fortes Aragão, Daniel Damous Dias, Lucas Henrique da Silva e Silva, Lúcia Aline Moura Reis, Hanna Carolina Farias Reis, Liliane Leal das Chagas, and et al. 2024. "Sequencing and Description of the Mitochondrial Genome of Orthopodomyia fascipes (Diptera: Culicidae)" Genes 15, no. 7: 874. https://doi.org/10.3390/genes15070874
APA StyleSilva, F. S. d., Nascimento, B. L. S. d., Cruz, A. C. R., Silva, S. P. d., Aragão, C. F., Dias, D. D., Silva, L. H. d. S. e., Reis, L. A. M., Reis, H. C. F., Chagas, L. L. d., Rosa Jr., J. W., Vieira, D. B. R., Brandão, R. C. F., Medeiros, D. B. d. A., & Nunes Neto, J. P. (2024). Sequencing and Description of the Mitochondrial Genome of Orthopodomyia fascipes (Diptera: Culicidae). Genes, 15(7), 874. https://doi.org/10.3390/genes15070874