

Ubiquitination-Related Gene Signature, Nomogram and Immune Features for Prognostic Prediction in Patients with Head and Neck Squamous Cell Carcinoma


Abstract
:1. Introduction
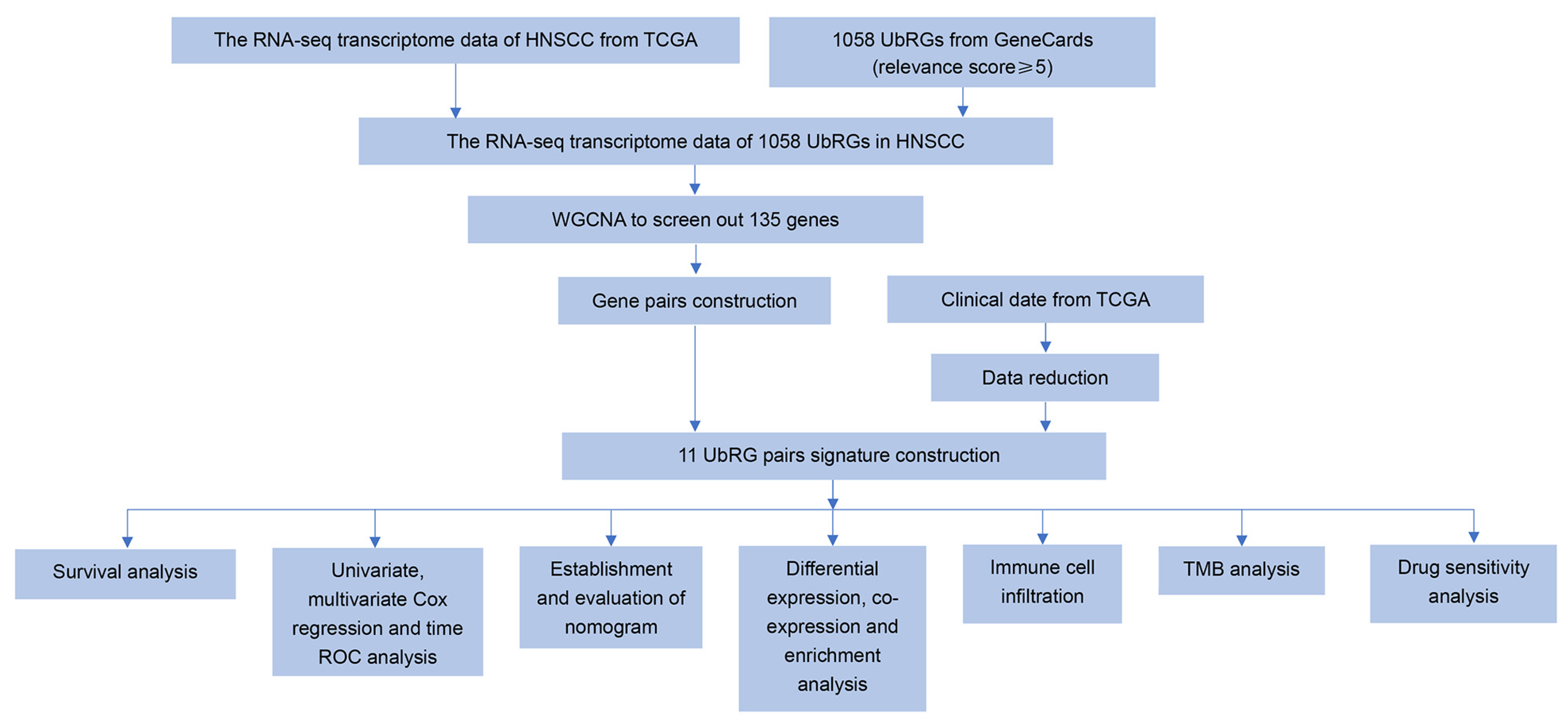
2. Materials and Methods
2.1. Collection of UbRGs and Sample Data
2.2. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.3. Ubiquitination-Related Gene Signature Construction in HNSCC
2.4. Verification of the Prognostic UbRG Signature
2.5. Development and Validation of the Nomogram
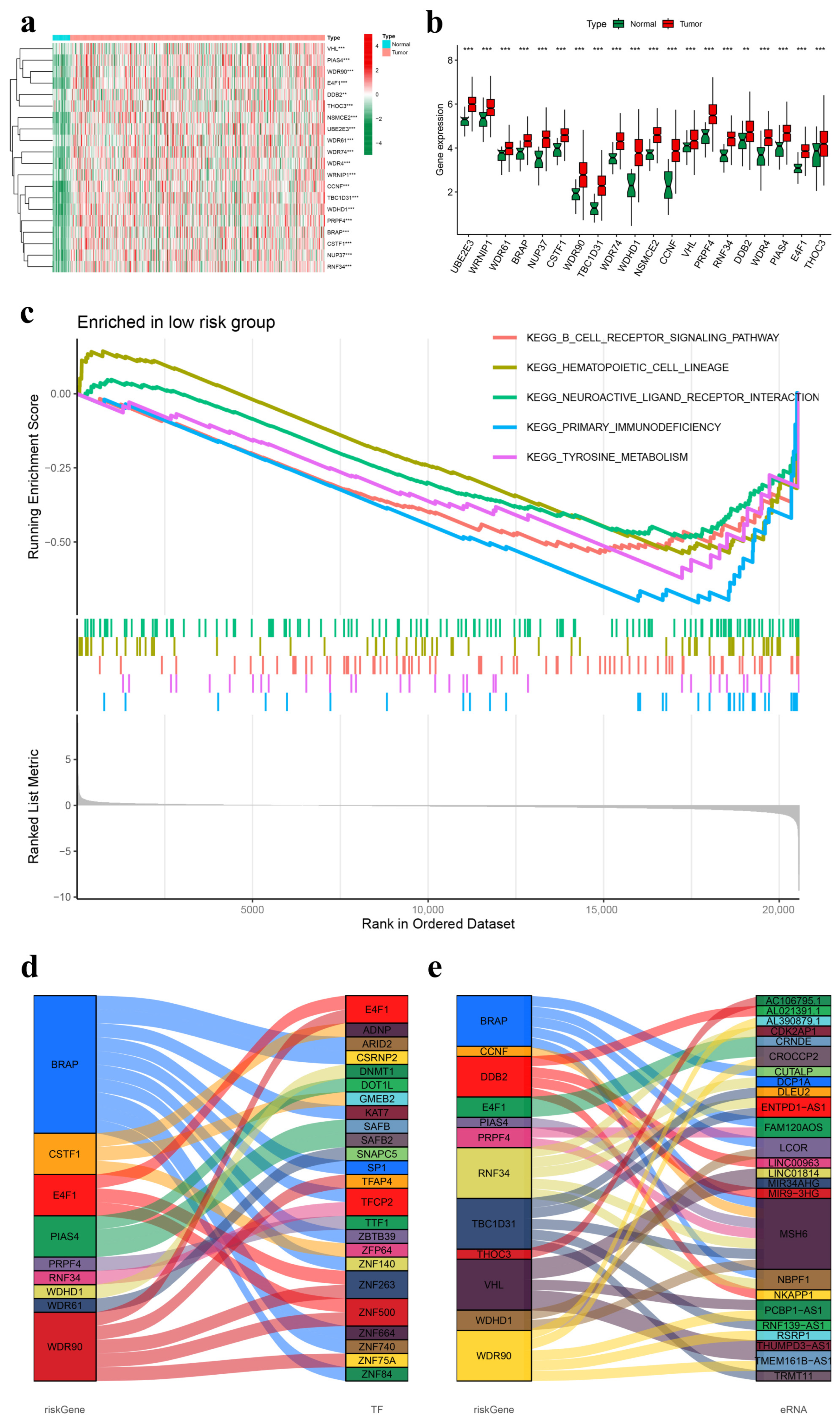
2.6. Gene Differential Expression Analysis of the Prognostic UbRG Signature
2.7. Gene Set Enrichment Analysis (GSEA)
2.8. Construction of Functional Interaction Networks of Model Genes, Transcription Factors, and Enhancer RNAs
2.9. Immune Landscapes Related to the Signature
2.10. Tumor Mutation Burden (TMB) Analysis
2.11. Drug Sensitivity Prediction
2.12. Statistical Methods
3. Results
3.1. Co-Expression Network of Ubiquitination-Related Genes
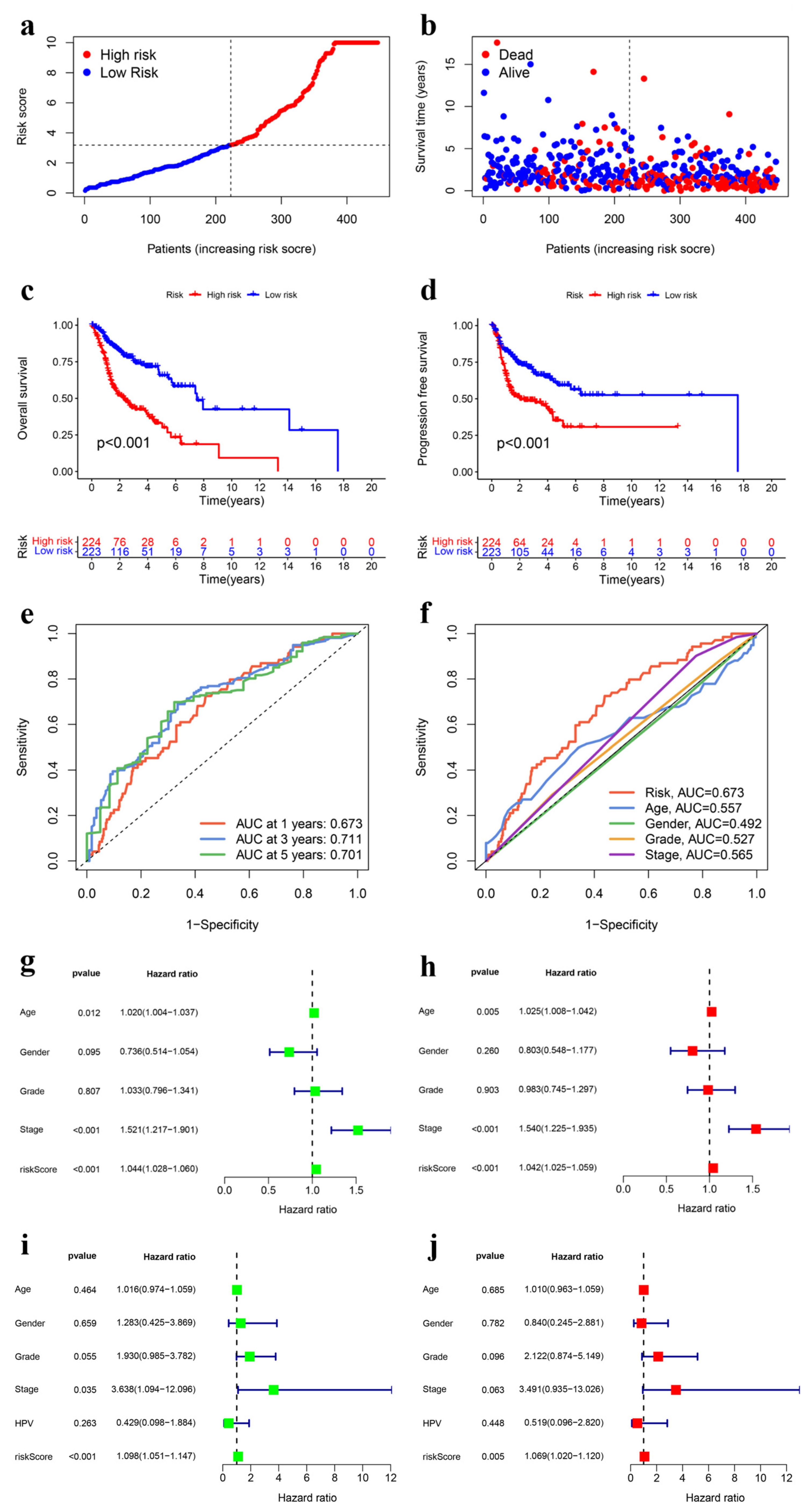
3.2. Construction and Internal Validation of the UbRG Signature
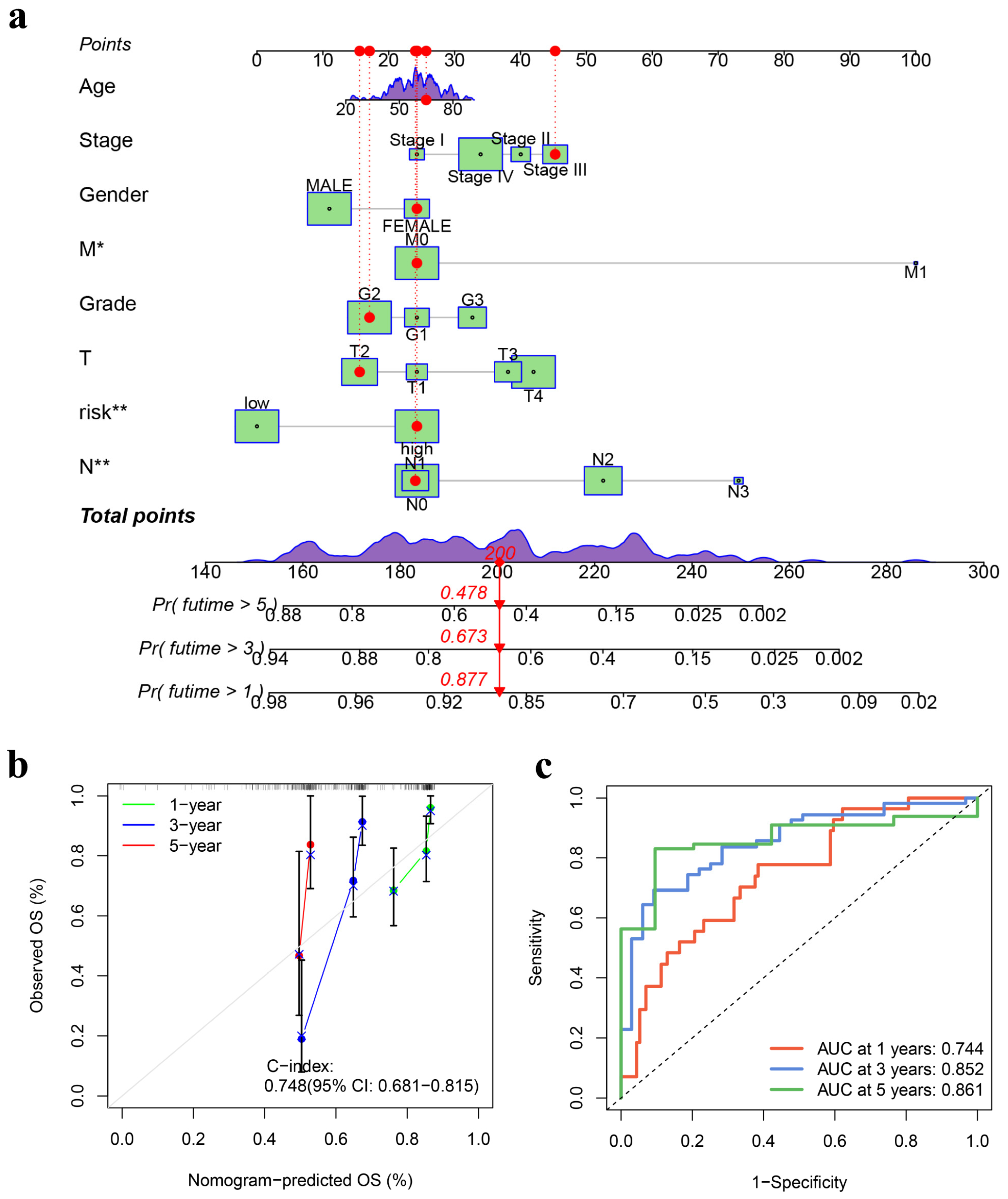
3.3. Development and Validation of a Prognostic Nomogram
3.4. External Analysis of the UbRG Signature
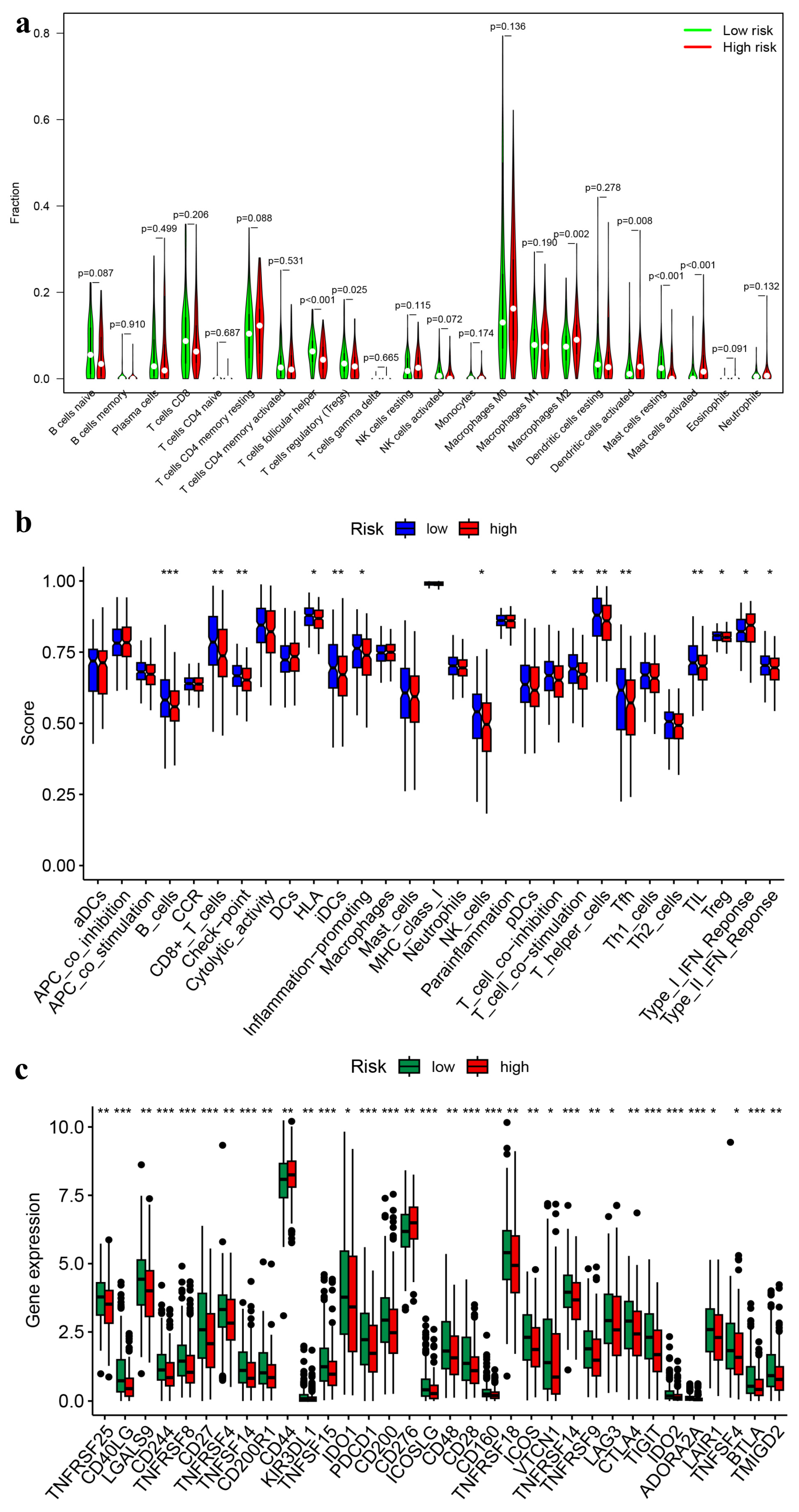
3.5. Immune Landscapes Related to the Signature
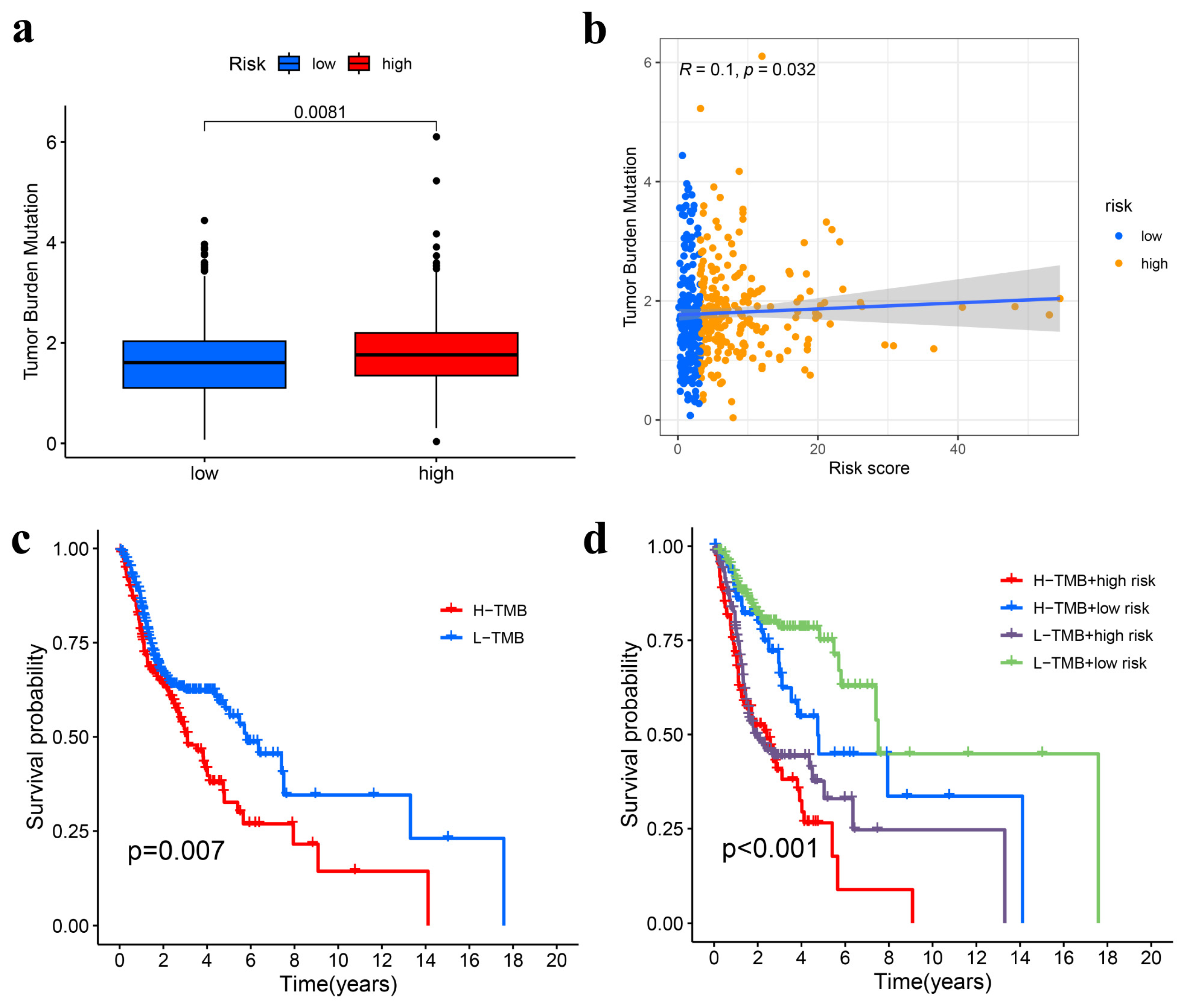
3.6. TMB Analysis
3.7. Drug Responses in High- and Low-Risk Groups of HNSCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Huang, X.; Duijf, P.H.G.; Sriram, S.; Perera, G.; Vasani, S.; Kenny, L.; Leo, P.; Punyadeera, C. Circulating tumour DNA alterations: Emerging biomarker in head and neck squamous cell carcinoma. J. Biomed. Sci. 2023, 30, 65. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Aggarwal, S.K.; Verma, S.S.; Awasthee, N.; Dhasmana, A.; Aggarwal, S.; Das, S.N.; Nair, M.S.; Yadav, S.; Gupta, S.C. Epoxyazadiradione exhibit activities in head and neck squamous cell carcinoma by targeting multiple pathways. Apoptosis 2020, 25, 763–782. [Google Scholar] [CrossRef] [PubMed]
- Märkl, F.; Huynh, D.; Endres, S.; Kobold, S. Utilizing chemokines in cancer immunotherapy. Trends Cancer 2022, 8, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Szeto, G.L.; Finley, S.D. Integrative Approaches to Cancer Immunotherapy. Trends Cancer 2019, 5, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Ruffin, A.T.; Li, H.; Vujanovic, L.; Zandberg, D.P.; Ferris, R.L.; Bruno, T.C. Improving head and neck cancer therapies by immunomodulation of the tumour microenvironment. Nat. Rev. Cancer 2023, 23, 173–188. [Google Scholar] [CrossRef]
- Vos, J.L.; Elbers, J.B.W.; Krijgsman, O.; Traets, J.J.H.; Qiao, X.; van der Leun, A.M.; Lubeck, Y.; Seignette, I.M.; Smit, L.A.; Willems, S.M.; et al. Neoadjuvant immunotherapy with nivolumab and ipilimumab induces major pathological responses in patients with head and neck squamous cell carcinoma. Nat. Commun. 2021, 12, 7348. [Google Scholar] [CrossRef]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef]
- Yang, Y.; He, Y.; Wang, X.; Liang, Z.; He, G.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. Protein SUMOylation modification and its associations with disease. Open Biol. 2017, 7, 170167. [Google Scholar] [CrossRef] [PubMed]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- van Wijk, S.J.; Fulda, S.; Dikic, I.; Heilemann, M. Visualizing ubiquitination in mammalian cells. EMBO Rep. 2019, 20, e46520. [Google Scholar] [CrossRef] [PubMed]
- Cruz Walma, D.A.; Chen, Z.; Bullock, A.N.; Yamada, K.M. Ubiquitin ligases: Guardians of mammalian development. Nat. Rev. Mol. Cell Biol. 2022, 23, 350–367. [Google Scholar] [CrossRef]
- Lee, J.C.; Peter, M.E. Regulation of apoptosis by ubiquitination. Immunol. Rev. 2003, 193, 39–47. [Google Scholar] [CrossRef]
- Park, H.B.; Kim, J.W.; Baek, K.H. Regulation of Wnt Signaling through Ubiquitination and Deubiquitination in Cancers. Int. J. Mol. Sci. 2020, 21, 3904. [Google Scholar] [CrossRef]
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37, 937–953. [Google Scholar] [CrossRef]
- Han, C.; Yang, L.; Choi, H.H.; Baddour, J.; Achreja, A.; Liu, Y.; Li, Y.; Li, J.; Wan, G.; Huang, C.; et al. Amplification of USP13 drives ovarian cancer metabolism. Nat. Commun. 2016, 7, 13525. [Google Scholar] [CrossRef]
- Yang, Y.; Staudt, L.M. Protein ubiquitination in lymphoid malignancies. Immunol. Rev. 2015, 263, 240–256. [Google Scholar] [CrossRef]
- Hou, Y.C.; Deng, J.Y. Role of E3 ubiquitin ligases in gastric cancer. World J. Gastroenterol. 2015, 21, 786–793. [Google Scholar] [CrossRef]
- Behl, T.; Chadha, S.; Sachdeva, M.; Kumar, A.; Hafeez, A.; Mehta, V.; Bungau, S. Ubiquitination in rheumatoid arthritis. Life Sci. 2020, 261, 118459. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, L.; Zeng, C.; Long, H.; Duan, G.; Yin, G.; Dai, X.; Lin, Z. E3 Ubiquitin Ligase HRD1 Promotes Lung Tumorigenesis by Promoting Sirtuin 2 Ubiquitination and Degradation. Mol. Cell Biol. 2020, 40, e00257-00219. [Google Scholar] [CrossRef]
- Bai, J.; Zhou, Y.; Chen, G.; Zeng, J.; Ding, J.; Tan, Y.; Zhou, J.; Li, G. Overexpression of Cullin1 is associated with poor prognosis of patients with gastric cancer. Hum. Pathol. 2011, 42, 375–383. [Google Scholar] [CrossRef]
- Chen, X.; Chen, S.; Jiang, Z.; Gong, Q.; Tang, D.; Luo, Q.; Liu, X.; He, S.; He, A.; Wu, Y.; et al. Ubiquitination-Related miRNA-mRNA Interaction Is a Potential Mechanism in the Progression of Retinoblastoma. Investig. Ophthalmol. Vis. Sci. 2021, 62, 3. [Google Scholar] [CrossRef]
- Deng, Z.M.; Hu, W.; Dai, F.F.; Yuan, M.Q.; Hu, M.; Cheng, Y.X. Immune-Related Genes to Construct a Novel Prognostic Model of Breast Cancer: A Chemosensitivity-Based Study. Front. Immunol. 2021, 12, 734745. [Google Scholar] [CrossRef] [PubMed]
- Iasonos, A.; Schrag, D.; Raj, G.V.; Panageas, K.S. How to build and interpret a nomogram for cancer prognosis. J. Clin. Oncol. 2008, 26, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Vučićević, D.; Corradin, O.; Ntini, E.; Scacheri, P.C.; Ørom, U.A. Long ncRNA expression associates with tissue-specific enhancers. Cell Cycle 2015, 14, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Jiang, Z.; Chen, C.; Wang, X. Classification of triple-negative breast cancers based on Immunogenomic profiling. J. Exp. Clin. Cancer Res. 2018, 37, 327. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, L.; Liu, M.; Li, Y.; Sun, Q. Novel immune-related prognostic model and nomogram for breast cancer based on ssGSEA. Front. Genet. 2022, 13, 957675. [Google Scholar] [CrossRef] [PubMed]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef] [PubMed]
- Gunassekaran, G.R.; Poongkavithai Vadevoo, S.M.; Baek, M.C.; Lee, B. M1 macrophage exosomes engineered to foster M1 polarization and target the IL-4 receptor inhibit tumor growth by reprogramming tumor-associated macrophages into M1-like macrophages. Biomaterials 2021, 278, 121137. [Google Scholar] [CrossRef]
- Han, S.; Bao, X.; Zou, Y.; Wang, L.; Li, Y.; Yang, L.; Liao, A.; Zhang, X.; Jiang, X.; Liang, D.; et al. d-lactate modulates M2 tumor-associated macrophages and remodels immunosuppressive tumor microenvironment for hepatocellular carcinoma. Sci. Adv. 2023, 9, eadg2697. [Google Scholar] [CrossRef]
- Tang, T.; Cheng, X.; Truong, B.; Sun, L.; Yang, X.; Wang, H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol. Ther. 2021, 219, 107709. [Google Scholar] [CrossRef]
- Slebioda, T.J.; Rowley, T.F.; Ferdinand, J.R.; Willoughby, J.E.; Buchan, S.L.; Taraban, V.Y.; Al-Shamkhani, A. Triggering of TNFRSF25 promotes CD8⁺ T-cell responses and anti-tumor immunity. Eur. J. Immunol. 2011, 41, 2606–2611. [Google Scholar] [CrossRef]
- Zhao, B.; Li, H.; Xia, Y.; Wang, Y.; Wang, Y.; Shi, Y.; Xing, H.; Qu, T.; Wang, Y.; Ma, W. Immune checkpoint of B7-H3 in cancer: From immunology to clinical immunotherapy. J. Hematol. Oncol. 2022, 15, 153. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Yang, C.; Gao, F. The state of CD44 activation in cancer progression and therapeutic targeting. FEBS J. 2022, 289, 7970–7986. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Bell, R.B.; Bifulco, C.B.; Burtness, B.; Gillison, M.L.; Harrington, K.J.; Le, Q.T.; Lee, N.Y.; Leidner, R.; Lewis, R.L.; et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J. Immunother. Cancer 2019, 7, 184. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, Z.; Xia, Y.; Li, J.; Wu, Y.; Wang, Y.; Jiang, H.; Cheng, J. Integrative Single-Cell and Bulk Transcriptomes Analyses Identify Intrinsic HNSCC Subtypes with Distinct Prognoses and Therapeutic Vulnerabilities. Clin. Cancer Res. 2023, 29, 2845–2858. [Google Scholar] [CrossRef] [PubMed]
- König, I.R.; Fuchs, O.; Hansen, G.; von Mutius, E.; Kopp, M.V. What is precision medicine? Eur. Respir. J. 2017, 50, 1700391. [Google Scholar] [CrossRef]
- Li, J.; Dai, Y.; Ge, H.; Guo, S.; Zhang, W.; Wang, Y.; Liu, L.; Cheng, J.; Jiang, H. The deubiquitinase USP7 promotes HNSCC progression via deubiquitinating and stabilizing TAZ. Cell Death Dis. 2022, 13, 677. [Google Scholar] [CrossRef]
- Wang, F.; Wang, J.; Wang, J.; Zhang, L.; Fu, H.; Li, J.; Tian, T.; Zuo, J.; Lv, W.; Ma, X. BCCIPβ facilitates p53 ubiquitination via binding with E6 protein in high-risk HPV positive head and neck squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2020, 529, 685–691. [Google Scholar] [CrossRef]
- Nanayakkara, D.M.; Nguyen, M.N.; Wood, S.A. Deubiquitylating enzyme, USP9X, regulates proliferation of cells of head and neck cancer lines. Cell Prolif. 2016, 49, 494–502. [Google Scholar] [CrossRef]
- Guo, Y.; Wu, Z.; Cen, K.; Bai, Y.; Dai, Y.; Mai, Y.; Hong, K.; Qu, L. Establishment and validation of a ubiquitination-related gene signature associated with prognosis in pancreatic duct adenocarcinoma. Front. Immunol. 2023, 14, 1171811. [Google Scholar] [CrossRef]
- Zuo, H.; Chen, L.; Li, N.; Song, Q. Identification of a Ubiquitination-Related Gene Risk Model for Predicting Survival in Patients with Pancreatic Cancer. Front. Genet. 2020, 11, 612196. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Deng, W.; Chen, P.; Mao, Q.; Chen, H.; Zhuo, Z.; Huang, Z.; Chen, K.; Huang, J.; Luo, Y. Development and validation of a novel ubiquitination-related gene prognostic signature based on tumor microenvironment for colon cancer. Transl. Cancer Res. 2022, 11, 3724–3740. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Gong, J. Prognostic Signature, Immune Features, and Therapeutic Responses of a Novel Ubiquitination-Related Gene Signature in Lung Adenocarcinoma. J. Oncol. 2022, 2022, 2524649. [Google Scholar] [CrossRef] [PubMed]
- Bommi, P.V.; Chand, V.; Mukhopadhyay, N.K.; Raychaudhuri, P.; Bagchi, S. NER-factor DDB2 regulates HIF1α and hypoxia-response genes in HNSCC. Oncogene 2020, 39, 1784–1796. [Google Scholar] [CrossRef] [PubMed]
- Bommi, P.V.; Ravindran, S.; Raychaudhuri, P.; Bagchi, S. DDB2 regulates Epithelial-to-Mesenchymal Transition (EMT) in Oral/Head and Neck Squamous Cell Carcinoma. Oncotarget 2018, 9, 34708–34718. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, K.; Chen, J.; Wang, X.; Ling, R.; Cheng, M.; Chen, Z.; Chen, F.; He, Q.; Li, S.; et al. Aberrant translation regulated by METTL1/WDR4-mediated tRNA N7-methylguanosine modification drives head and neck squamous cell carcinoma progression. Cancer Commun. 2022, 42, 223–244. [Google Scholar] [CrossRef]
- Plafker, K.S.; Zyla, K.; Berry, W.; Plafker, S.M. Loss of the ubiquitin conjugating enzyme UBE2E3 induces cellular senescence. Redox Biol. 2018, 17, 411–422. [Google Scholar] [CrossRef]
- Priest, C.; Nagari, R.T.; Bideyan, L.; Lee, S.D.; Nguyen, A.; Xiao, X.; Tontonoz, P. Brap regulates liver morphology and hepatocyte turnover via modulation of the Hippo pathway. Proc. Natl. Acad. Sci. USA 2022, 119, e2201859119. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef]
- Zhou, L.; Cheng, Q.; Hu, Y.; Tan, H.; Li, X.; Wu, S.; Zhou, T.; Zhou, J. Cuproptosis-related LncRNAs are potential prognostic and immune response markers for patients with HNSCC via the integration of bioinformatics analysis and experimental validation. Front. Oncol. 2022, 12, 1030802. [Google Scholar] [CrossRef]
- Zhang, L.; Li, B.; Peng, Y.; Wu, F.; Li, Q.; Lin, Z.; Xie, S.; Xiao, L.; Lin, X.; Ou, Z.; et al. The prognostic value of TMB and the relationship between TMB and immune infiltration in head and neck squamous cell carcinoma: A gene expression-based study. Oral. Oncol. 2020, 110, 104943. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.L.; Yang, K.B.; Xu, C.; Feng, R.J.; Li, W.F.; Ma, J. Development of a prediction model for radiotherapy response among patients with head and neck squamous cell carcinoma based on the tumor immune microenvironment and hypoxia signature. Cancer Med. 2022, 11, 4673–4687. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiu, J.; Farrell, A.; Baca, Y.; Arai, H.; Battaglin, F.; Kawanishi, N.; Soni, S.; Zhang, W.; Millstein, J.; et al. Mutational analysis of microsatellite-stable gastrointestinal cancer with high tumour mutational burden: A retrospective cohort study. Lancet Oncol. 2023, 24, 151–161. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.I.; Seiwert, T.Y.; Chow, L.Q.M.; Gupta, S.; Weiss, J.; Gluck, I.; Eder, J.P.; Burtness, B.; Tahara, M.; Keam, B.; et al. Influence of tumor mutational burden, inflammatory gene expression profile, and PD-L1 expression on response to pembrolizumab in head and neck squamous cell carcinoma. J. Immunother. Cancer 2022, 10, e003026. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, N.; Sento, S.; Yoshizawa, Y.; Sasabe, E.; Kudo, Y.; Yamamoto, T. Current Trends and Future Prospects of Molecular Targeted Therapy in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 22, 240. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviations | Full Names |
|---|---|
| UBE2E3 | Ubiquitin-conjugating enzyme E2 E3 |
| WRNIP1 | Werner helicase-interacting protein 1 |
| WDR61 | WD repeat domain 61 |
| BRAP | BRCA1-associated protein |
| NUP37 | Nucleoporin 37 |
| CSTF1 | Cleavage stimulation factor 1 |
| WDR90 | WD repeat domain 90 |
| TBC1D31 | TBC1 domain family member 31 |
| WDR74 | WD repeat domain 74 |
| WDHD1 | WD repeat and high-mobility group box DNA binding protein 1 |
| NSMCE2 | NSE2 (MMS21) homolog, SMC5-SMC6 complex SUMO ligase |
| CCNF | Cyclin F |
| VHL | Von Hippel−Lindau tumor suppressor |
| PRPF4 | Pre-mRNA splicing tri-snRNP complex factor PRPF4 |
| RNF34 | Ring finger protein 34 |
| DDB2 | Damage specific DNA binding protein 2 |
| WDR4 | WD repeat domain 4 |
| PIAS4 | Protein inhibitor of activated STAT 4 |
| E4F1 | E4F transcription factor 1 |
| THOC3 | THO complex subunit 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Zhou, L.; Shi, M.; Yu, J.; Xie, Y.; Sun, Y. Ubiquitination-Related Gene Signature, Nomogram and Immune Features for Prognostic Prediction in Patients with Head and Neck Squamous Cell Carcinoma. Genes 2024, 15, 880. https://doi.org/10.3390/genes15070880
Yang H, Zhou L, Shi M, Yu J, Xie Y, Sun Y. Ubiquitination-Related Gene Signature, Nomogram and Immune Features for Prognostic Prediction in Patients with Head and Neck Squamous Cell Carcinoma. Genes. 2024; 15(7):880. https://doi.org/10.3390/genes15070880
Chicago/Turabian StyleYang, Huiwen, Liuqing Zhou, Mengwen Shi, Jintao Yu, Yi Xie, and Yu Sun. 2024. "Ubiquitination-Related Gene Signature, Nomogram and Immune Features for Prognostic Prediction in Patients with Head and Neck Squamous Cell Carcinoma" Genes 15, no. 7: 880. https://doi.org/10.3390/genes15070880