
Comparative Analysis and Phylogeny of the Complete Chloroplast Genomes of Nine Cynanchum (Apocynaceae) Species
Abstract
:1. Introduction
2. Results
2.1. Genome Size and Features
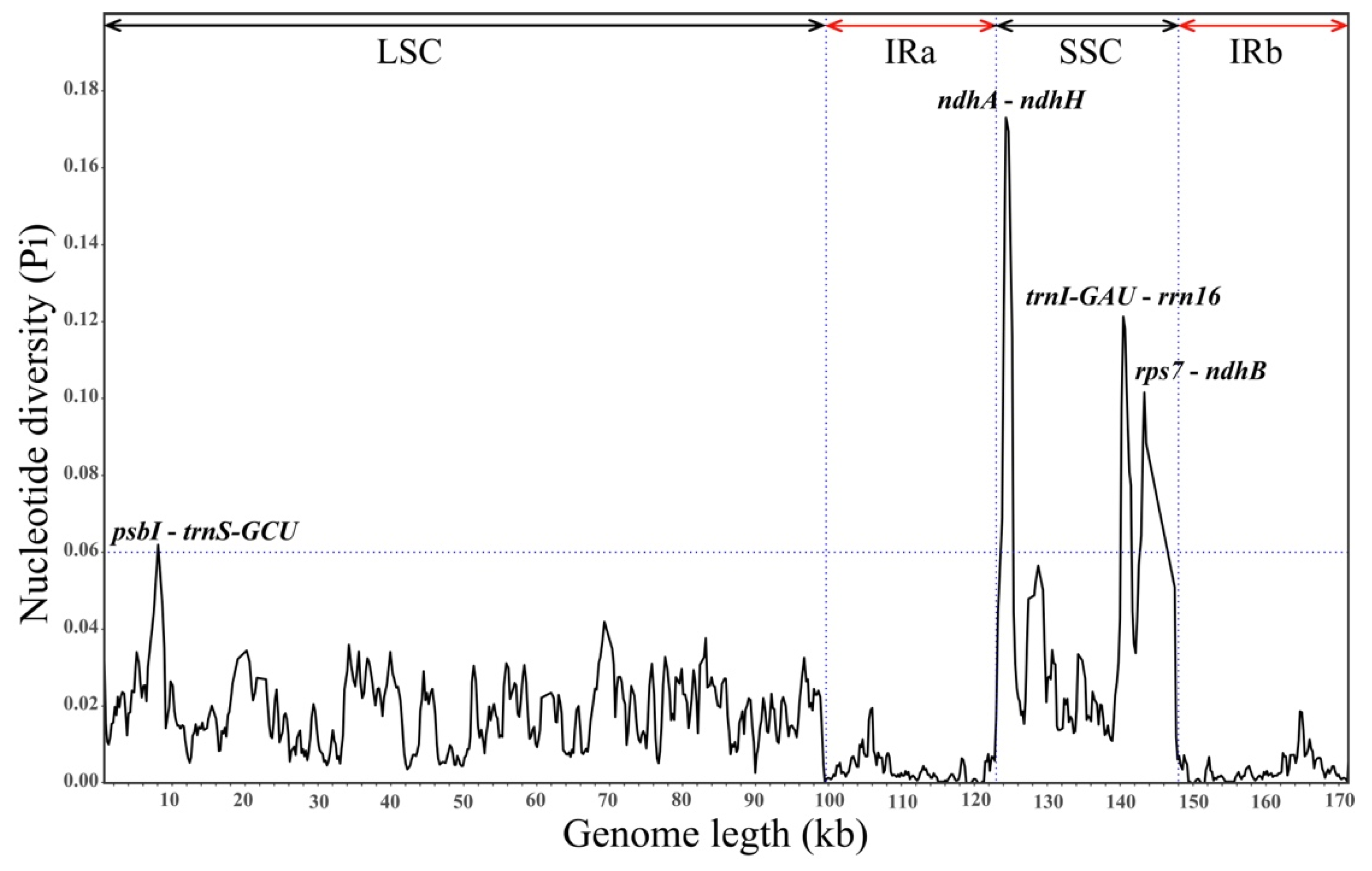
2.2. Comparative Genomics and Divergence Hotspots
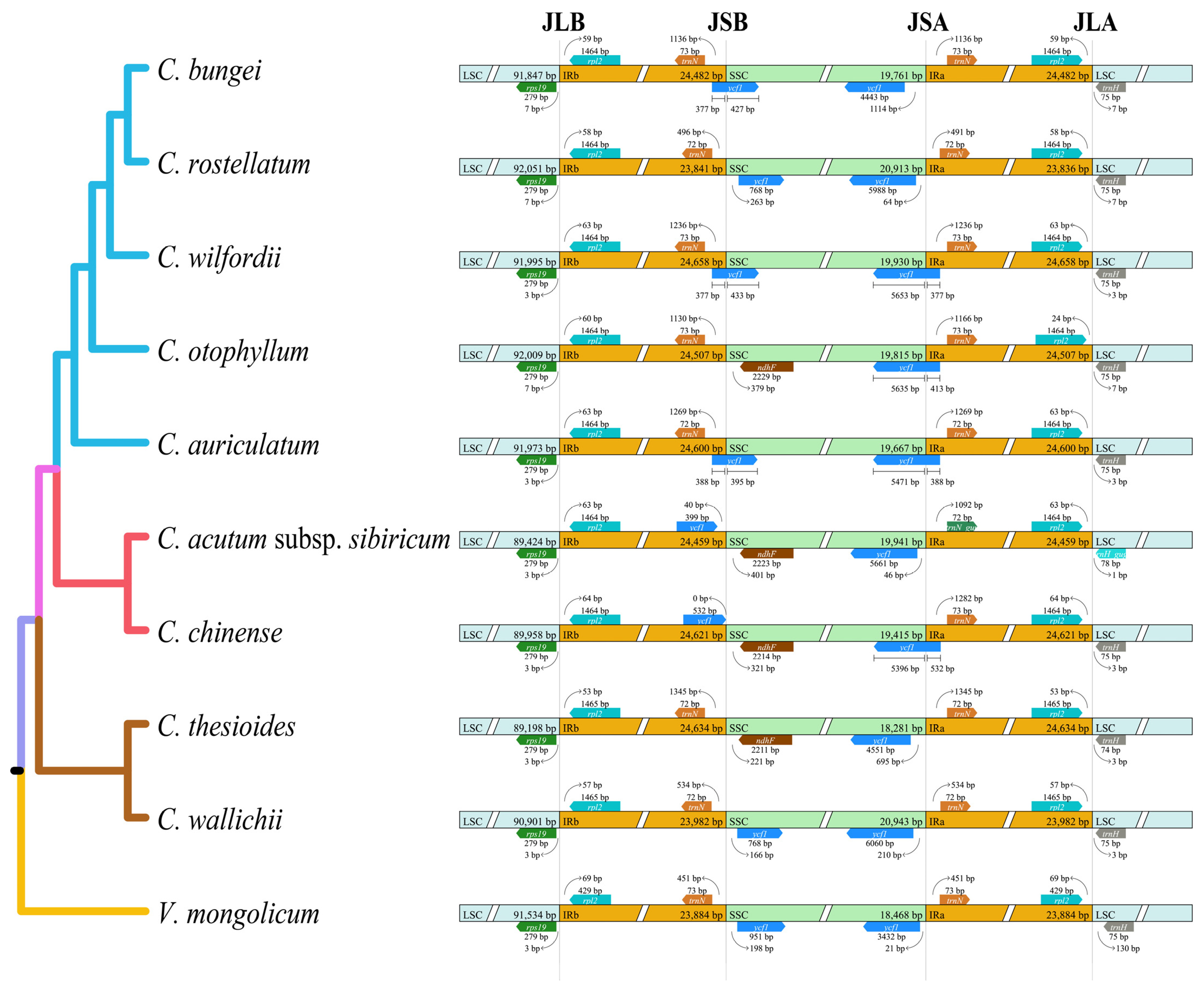
2.3. Boundaries between IR and SC Regions
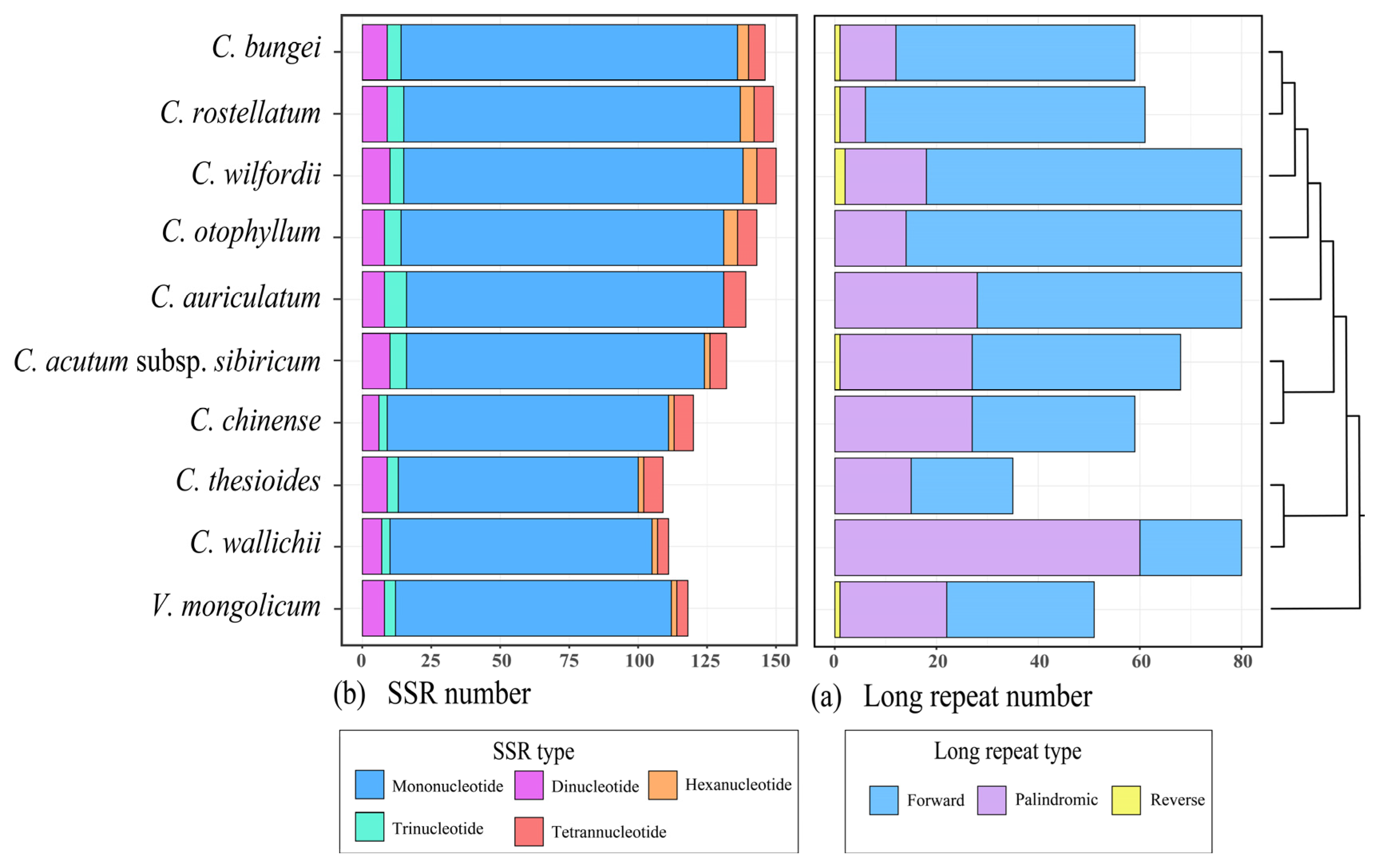
2.4. Repeat Identification
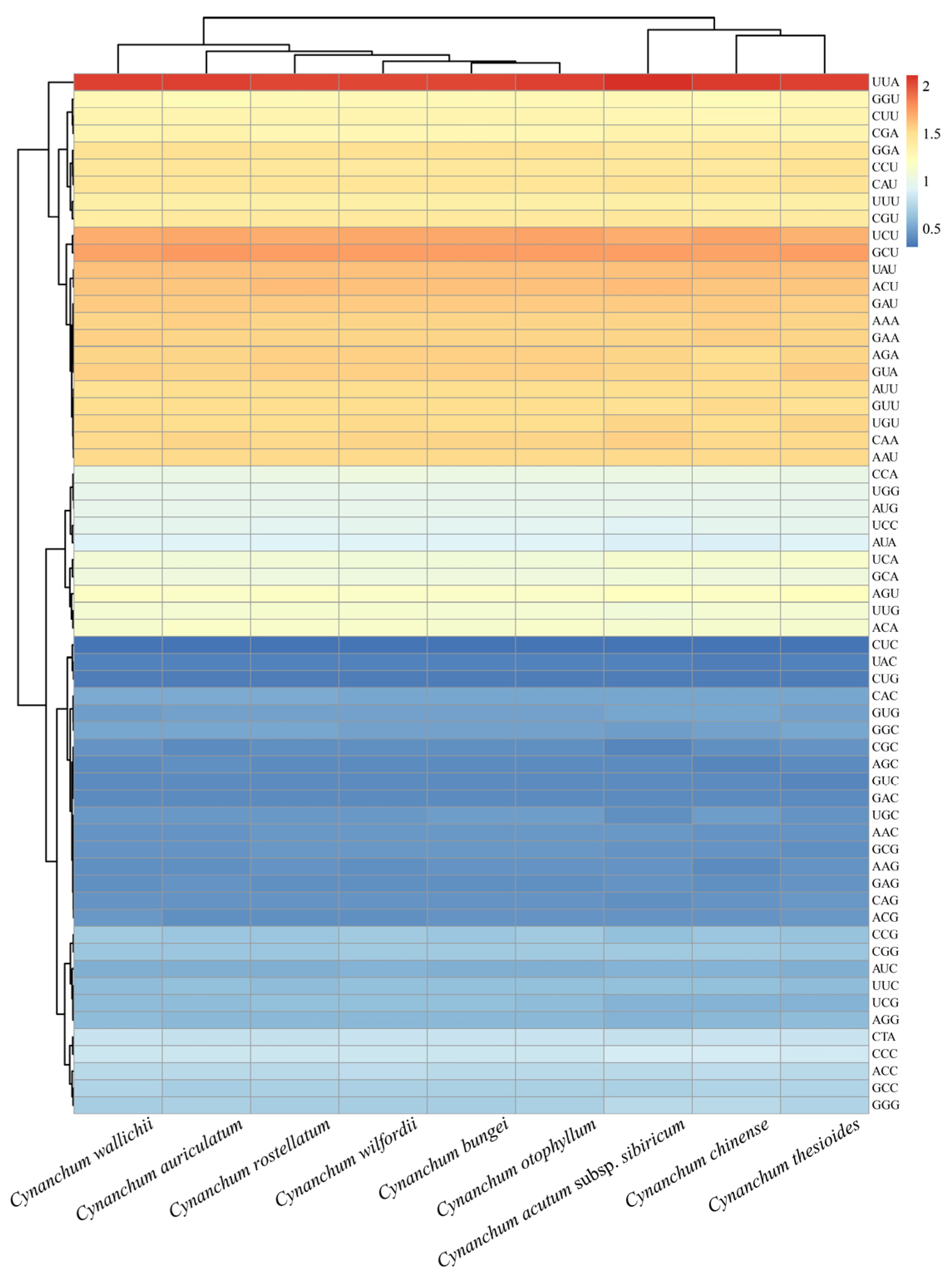
2.5. Codon Usage Analysis
2.6. Phylogenetic Analyses and Divergence Time Estimation
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Taxon Sampling, DNA Extraction, and PCG Sequencing
5.2. Chloroplast Genome and Annotation
5.3. Comparative Genomics and Structural Analyses
5.4. Repeat Sequence and Codon Preference Analyses
5.5. Phylogenetic Inference and Divergence Time Estimation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, P.T.; Michael, G.G.; Stevens, W.D. Asclepiadaceae. In Flora of China; Wu, Z., Raven, P., Eds.; Science Press: Beijing, China, 1995; Volume 16, pp. 311–313. [Google Scholar]
- Endress, M.E.; Liede-Schumann, S.; Meve, U. An updated classification for Apocynaceae. Phytotaxa 2014, 159, 115. [Google Scholar] [CrossRef]
- Yang, J.; Qing, Z.; Wang, D.; Yang, H. Distribution of medicinal plant resources of Cynanchum in Guizhou. Guizhou Sci. 2018, 36, 50–52. [Google Scholar]
- Zhang, X.; Yang, Z.; Li, Z.; Zhang, F.; Hao, L. De novo transcriptome assembly and co-expression network analysis of Cynanchum thesioides: Identification of genes involved in resistance to drought stress. Gene 2019, 710, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.Y.; Yang, Z.R.; Zhang, X.Y.; Zhang, F.L.; Huang, X.M.; Han, X. Transcriptome-wide identification of WRKY transcription factors and their expression profiles under different stress in Cynanchum thesioides. PeerJ 2022, 10, e144362022. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qu, J.; Yu, S.S.; Hu, Y.C.; Huang, X.Z. Seven new steroidal glycosides from the roots of Cynanchum forrestii. Steroids 2007, 72, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, I.; Sen, O.; Erenler, R.; Demirtas, I.; Behcet, L. Behcet bioactivity-guided isolation of flavonoids from Cynanchum acutum L. subsp. sibiricum (willd.) Rech. f. and investigation of their antiproliferative activity. Nat. Prod. Res. 2017, 31, 2629–2633. [Google Scholar]
- Wei, L.; Yang, M.; Huang, L.; Li, J.L. Antibacterial and antioxidant flavon-oid derivatives from the fruits of Metaplexis japonica. Food Chem. 2019, 289, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Jamarkattel-Pandit, N.; Kim, H. Neuroprotective effect of Metaplexis japonica against in vitro ischemia model. J. Health Allied Sci. 2019, 3, 51–55. [Google Scholar] [CrossRef]
- Choi, J.H.; Jung, B.H.; Kang, O.H.; Choi, H.J.; Park, P.S.; Cho, S.H.; Kim, Y.C.; Sohn, D.H.; Park, H.; Lee, J.H.; et al. The anti-inflammatory and anti-nociceptive effects of ethyl acetate fraction of Cynanchi paniculati radix. Biol. Pharm. Bull. 2006, 29, 971–975. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, J.; Liang, Z.; Zhao, C. New lignans and their biological activities. Chem. Biodivers. 2014, 11, 1–54. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, B.M.; Liu, C.H.; Liu, H.X.; Liu, A.X. First report of Sclerotium delphinii on Cynanchum Paniculatum. Australas. Plant Dis. Notes 2017, 12, 1–4. [Google Scholar] [CrossRef]
- Lee, S.K.; Nam, K.A.; Heo, Y.H. Cytotoxic activity and G2/M cell cycle arrestmediated by antofine, a phenanthroindolizidine alkaloid isolated from Cynanchum paniculatum. Planta Medica 2003, 69, 21–25. [Google Scholar] [CrossRef]
- Dou, J.; Li, P.; Bi, Z.M.; Zhou, J.L. New C21 steroidal glycoside from Cynanchum paniculatum. Chin. Chem. Lett. 2007, 18, 300–302. [Google Scholar] [CrossRef]
- Min, H.Y.; Chung, H.J.; Kim, E.H.; Kim, S.; Park, E.J.; Lee, S.K. Inhibition of cell growth and potentiation of tumor necrosis factor-α(TNF-α)- induced apoptosis by a phenanthroindolizidine alkaloid antofine in human colon cancer cells. Biochem. Pharmacol. 2010, 80, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.; Stevens, W.; and Li, P.T. Notes on the Asclepiadaceae of China. Novon 1995, 5, 1–16. [Google Scholar] [CrossRef]
- Liede-Schumann, S. Subtribe Astephaninae (Apocynaceae-Asclepiadoideae) reconsidered: New evidence based on cpDNA spacers. Ann. Mo. Bot. Gard. 2000, 88, 657–668. [Google Scholar] [CrossRef]
- Yamashiro, T.; Fukuda, T.; Yokoyama, J.; Maki, M. Molecular phylogeny of Vincetoxicum (Apocynaceae-Asclepiadoideae) based on the nucleotide sequences of cpDNA and nrDNA. Mol. Phylogenetics Evol. 2004, 31, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, M.; Livshultz, T.; Straub, S.C.K.; Simões, A.O.; Boutte, J.; McDonnell, A.; Foote, A. Evolution on the backbone: Apocynaceae phylogenomics and new perspectives on growth forms, flowers, and fruits. Am. J. Bot. 2018, 105, 495–513. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Jin, S.; Zhu, X.G.; Gitzendanner, M.A.; Soltis, D.E.; Soltis, P.S. Green giant-a tiny chloroplast genome with mighty power to produce high-value proteins: History and phylogeny. Plant Biotechnol. J. 2021, 19, 430–447. [Google Scholar] [CrossRef]
- Qin, H.H.; Cai, J.; Liu, C.K.; Zhou, R.X.; Price, M.; Zhou, S.D.; He, X.J. The plastid genome of twenty-two species from Ferula, Talassia, and Soranthus: Comparative analysis, phylogenetic implications, and adaptive evolution. BMC Plant Biol. 2023, 23, 9. [Google Scholar] [CrossRef]
- Yang, Z.; Ma, W.; Yang, X.; Wang, L.; Zhao, T.; Liang, L.; Wang, G.; Ma, Q. Plastome phylogenomics provide new perspective into the phylogeny and evolution of Betulaceae (Fagales). BMC Plant Biol. 2022, 22, 611. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.J.; Su, N.; Zhang, L.; Tong, R.C.; Zhang, X.H.; Wang, J.R. Chloroplast genomes elucidate diversity, phylogeny, and taxonomy of Pulsatilla (Ranunculaceae). Sci. Rep. 2020, 10, 19781. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.Y.; Liu, J.Q.; Hao, G.Q.; Zhang, L.; Mao, K.S.; Wang, X.J.; Zhang, D.; Ma, T.; Hu, Q.J.; Al-Shehbaz, I.A.; et al. Plastome phylogeny and early diversification of Brassicaceae. BMC Genom. 2017, 18, 176. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Hu, Q.J.; Al-Shehbaz, I.A.; Luo, X.; Zeng, T.T.; Guo, X.Y.; Liu, J.Q. Species delimitation and interspecific relationships of the genus Orychophragmus (Brassicaceae) inferred from whole chloroplast genomes. Front. Plant Sci. 2016, 7, 1826. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xi, Z.X.; Wang, M.C.; Guo, X.Y.; Ma, T. Plastome phylogeny and lineage diversification of Salicaceae with focus on poplars and willows. Ecol. Evol. 2018, 8, 7817–7823. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.H.; Zhang, L.; Zhang, C.; Chang, H.; Xi, Z.X.; Xu, X.T. Comparative analysis of the complete chloroplast genomes of six threatened subgenus Gynopodium (Magnolia) species. BMC Genom. 2022, 23, 716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, D. The complete chloroplast genome sequence of Cynanchum forrestii Schltr. (Asclepiadaceae) and its phylogenetic analysis. Mitochondrial DNA Part B Resour. 2019, 4, 3675–3676. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jang, W.J.; Kim, E.; Kim, J.; Gong, H.G.; Kang, J.S.; Shim, H.; Park, J.Y.; Yang, T.J. The complete plastome of Cynanchum rostellatum (Apocynaceae), an indigenous plant in Korea. Mitochondrial DNA Part. B Resour. 2022, 7, 2035–2039. [Google Scholar] [CrossRef]
- Zhang, T.W.; Fang, Y.J.; Wang, X.M.; Deng, X.; Zhang, X.W.; Hu, S.N.; Yu, J. The complete chloroplast and mitochondrial genome sequences of Boea hygrometrica: Insights into the evolution of plant organellar genomes. PLoS ONE 2012, 7, e30531. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.X.; Zhi, L.Q.; Jia, Y.; Zhong, Q.Y.; Liu, Z.L.; Yue, M.; Li, Z.H. Nucleotide diversity and demographic history of Pinus bungeana, an endangered conifer species endemic in China. J. Syst. Evol. 2020, 58, 282–294. [Google Scholar] [CrossRef]
- Zhang, F.J.; Wang, T.; Shu, X.C.; Wang, N.; Zhuang, W.B.; Wang, Z. Complete chloroplast genomes and comparative analyses of L. chinensis, L. anhuiensis, and L. aurea (Amaryllidaceae). Int. J. Mol. Sci. 2020, 21, 5729. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.W.; Lin, C.X.; Lee, S.Y.; Mao, L.F.; Meng, K.K. Comparative analysis of complete Ilex (Aquifoliaceae) chloroplast genomes: Insights into evolutionary dynamics and phylogenetic relationships. BMC Genom. 2022, 23, 203. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Mohanan, P.; Yang, T.J. Super-barcoding of medicinal materials based on complete Plastid genomes and nuclear ribosomal DNA. Authentication Chin. Med. Mater. DNA Technol. 2023, 363–389. [Google Scholar]
- Park, H.S.; Jayakodi, M.; Lee, S.H.; Jeon, J.H.; Lee, H.O.; Park, J.Y.; Moon, B.C.; Kim, C.K.; Wing, R.A.; Newmaster, S.G.; et al. Mitochondrial plastid DNA can cause DNA barcoding paradox in plants. Sci. Rep. 2017, 7, 14910. [Google Scholar]
- Song, Y.; Wang, S.J.; Ding, Y.M.; Xu, J.; Li, M.F.; Zhu, S.F. Chloroplast genomic resource of Paris for species discrimination. Sci. Rep. 2017, 7, 3427. [Google Scholar] [CrossRef]
- Cheng, H.; Li, J.F.; Zhang, H.; Cai, B.H.; Gao, Z.H.; Qiao, Y.S. The complete chloroplast genome sequence of strawberry (Fragaria × ananassa Duch.) and comparison with related species of Rosaceae. PeerJ 2017, 5, e3919. [Google Scholar] [CrossRef] [PubMed]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef]
- Tyagi, S.; Jung, J.A.; Kim, J.S.; Won, S.Y. Comparative analysis of the complete chloroplast genome of mainland Aster spathulifolius and other Aster species. Plants 2020, 9, 568. [Google Scholar] [CrossRef]
- Wang, L.; Xiao, A.H.; Ma, L.Y.; Chen, F.J.; Sang, Z.Y.; Duan, J. Identification of Mag- nolia wufengensis (Magnoliaceae) cultivars using phenotypic traits, SSR and SRAP markers: Insights into breeding and conservation. Genet. Mol. Res. 2017, 16, gmr16019473. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef]
- Wang, J.; Qian, J.; Jiang, Y.; Chen, X.C.; Zheng, B.J.; Chen, S.L.; Yang, F.F.; Xu, Z.C.; Duan, B.Z. Comparative analysis of chloroplast genome and new insights into phylogenetic relationships of Polygonatum and Tribe Polygonateae. Front. Plant Sci. 2022, 13, 882189. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.Y.; Wang, J.; Pu, T.T.; Dong, J.J.; Guan, Q.; Qian, J.; Shi, L.C.; Duan, B.Z. Com- parative analysis of medicinal plant Isodon rubescens and its common adulterants based on chloroplast genome sequencing. Front. Plant Sci. 2022, 13, 1036277. [Google Scholar] [CrossRef]
- Firetti, F.; Zuntini, A.R.; Gaiarsa, J.W.; Oliveira, R.S.; Lohmann, L.G.; VanSluys, M.A. Complete chloroplast genome sequences contribute to plant species delimitation: A case study of the Anemopaegma species complex. Am. J. Bot. 2017, 104, 1493–1509. [Google Scholar] [CrossRef]
- Yu, X.Q.; Drew, B.T.; Yang, J.B.; Gao, L.M.; Li, D.Z. Comparative chloroplast genomes of eleven Schima (Theaceae) species: Insights into DNA barcoding and phylogeny. PLoS ONE 2017, 12, e0178026. [Google Scholar] [CrossRef]
- Allen, G.C.; Floresvergara, M.A.; Krasynanski, S.; Kumar, S.; Thompson, W.F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethy lammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Huang, D.I.; Cronk, Q.C. Plann: A command-line application for annotating plastome sequences. Appl. Plant Sci. 2015, 3, 1500026. [Google Scholar] [CrossRef]
- Löytynoja, A.; Goldman, N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 2008, 320, 1632–1635. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2020, 48, D84–D86. [Google Scholar] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19, i54–i62. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Delbarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6, DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8, A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P.; et al. MrBayes 3.2, Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree v1. 4; University of Edinburgh: Edinburgh, UK, 2012; Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 25 November 2018).
- Yang, Z.H. PAML 4, Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B.; Yang, Z. Inferring speciation times under an episodic molecular clock. Syst. Biol. 2007, 56, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Barba-Montoya, J.; Dos, R.M.; Yang, Z. Comparison of different strategies for using fossil calibrations to generate the time prior in Bayesian molecular clock dating. Mol. Phylogenetics Evol. 2017, 114, 386–400. [Google Scholar] [CrossRef]
- Neupane, S.; Lewis, P.O.; Dessein, S.; Hunter, S.; Sushil, P.; Frederic, L. Evolution of woody life form on tropical mountains in the tribe Spermacoceae (Rubiaceae). Am. J. Bot. 2017, 104, 419–438. [Google Scholar] [CrossRef] [PubMed]
- Collinson, M.; Manchester, S.; Wilde, V.; Hayes, P. Fruit and seed floras from exceptionally preserved biotas in the European Paleogene. Bull. Geosci. 2009, 85, 155–162. [Google Scholar] [CrossRef]
- Ribeiro, P.L.; Rapini, A.; Damascena, L.S.; Leilton, S.D.; Cássio, V.D.B. Plant diversification in the Espinhaço Range: Insights from the biogeography of Minaria (Apocynaceae). Taxon 2014, 63, 1253–1264. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | C. auriculatum | C. bungei | C. chinense | C. otophyllum | C. rostellatum | C. acutum subsp. sibiricum | C. thesioides | C. wallichii | C. wilfordii |
|---|---|---|---|---|---|---|---|---|---|
| Total cpDNA size (bp) | 160,840 bp | 160,572 bp | 158,615 bp | 160,874 bp | 160,641 bp | 158,283 bp | 156,747 | 159,808 bp | 161,241 bp |
| LSC size (bp) | 91,737 | 91,847 | 89,958 | 92,009 | 92,051 | 89,424 | 89,198 | 90,901 | 91,995 |
| SSC size (bp) | 19,667 | 19,761 | 19,415 | 19,815 | 20,922 | 19,713 | 18,281 | 20,943 | 19,930 |
| IR size (bp) | 24,600 | 24,482 | 24,621 | 24,507 | 23,834 | 24,573 | 24,634 | 23,982 | 24,658 |
| Number of genes | 132 | 131 | 132 | 131 | 132 | 129 | 132 | 133 | 132 |
| Protein-coding genes | 87 | 86 | 87 | 86 | 87 | 84 | 87 | 88 | 87 |
| rRNA genes | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| tRNA genes | 37 | 37 | 37 | 37 | 37 | 37 | 37 | 37 | 37 |
| LSC GC% | 35.57 | 36.1 | 35.53 | 35.64 | 36.08 | 36.15 | 36.24 | 36.15 | 35.62 |
| SSC GC% | 31.46 | 30.2 | 31.88 | 31.56 | 32.11 | 32.27 | 32.57 | 31.92 | 31.31 |
| IR GC% | 42.75 | 44.55 | 42.77 | 42.69 | 43.68 | 43.23 | 43.39 | 43.69 | 42.75 |
| GC content(%) | 37.8 | 37.8 | 37.8 | 37.8 | 37.8 | 37.9 | 38.1 | 37.9 | 37.8 |
| Accession Number | KT220734 | OK271106 | MW415427 | OQ587923 | OL689165 | OQ390041 | MW864598 | OQ198623 | KT220733 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, E.; Ma, X.; Guo, T.; Wu, Y.; Zhang, L. Comparative Analysis and Phylogeny of the Complete Chloroplast Genomes of Nine Cynanchum (Apocynaceae) Species. Genes 2024, 15, 884. https://doi.org/10.3390/genes15070884
Zhang E, Ma X, Guo T, Wu Y, Zhang L. Comparative Analysis and Phylogeny of the Complete Chloroplast Genomes of Nine Cynanchum (Apocynaceae) Species. Genes. 2024; 15(7):884. https://doi.org/10.3390/genes15070884
Chicago/Turabian StyleZhang, Erdong, Xueling Ma, Ting Guo, Yujie Wu, and Lei Zhang. 2024. "Comparative Analysis and Phylogeny of the Complete Chloroplast Genomes of Nine Cynanchum (Apocynaceae) Species" Genes 15, no. 7: 884. https://doi.org/10.3390/genes15070884