
Exploring Microorganisms Associated to Acute Febrile Illness and Severe Neurological Disorders of Unknown Origin: A Nanopore Metagenomics Approach
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
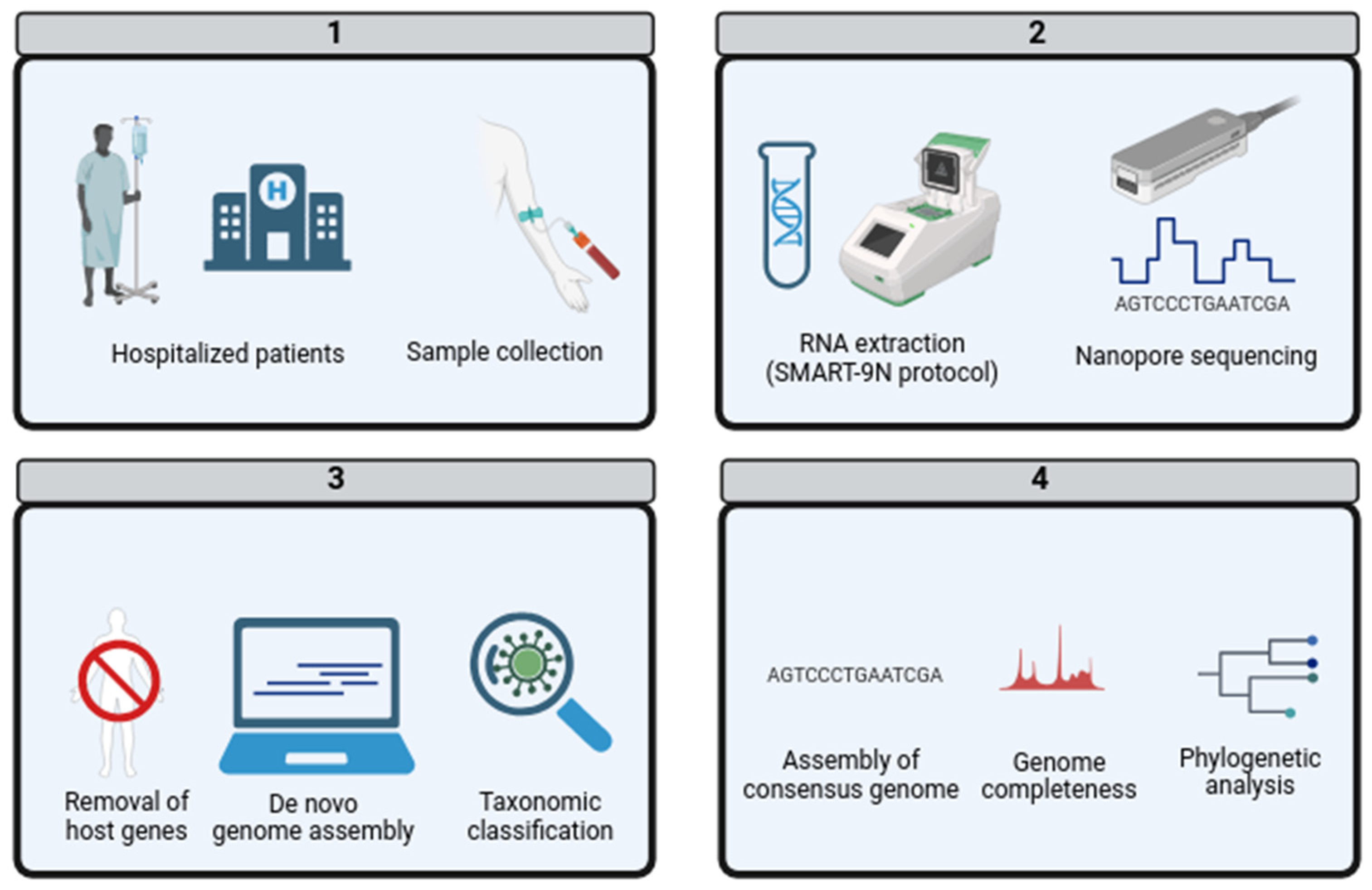
2. Materials and Methods
2.1. Clinical Sampling
2.2. RNA Extraction and Nanopore Sequencing
2.3. De Novo Genome Assembly and Taxonomic Classification
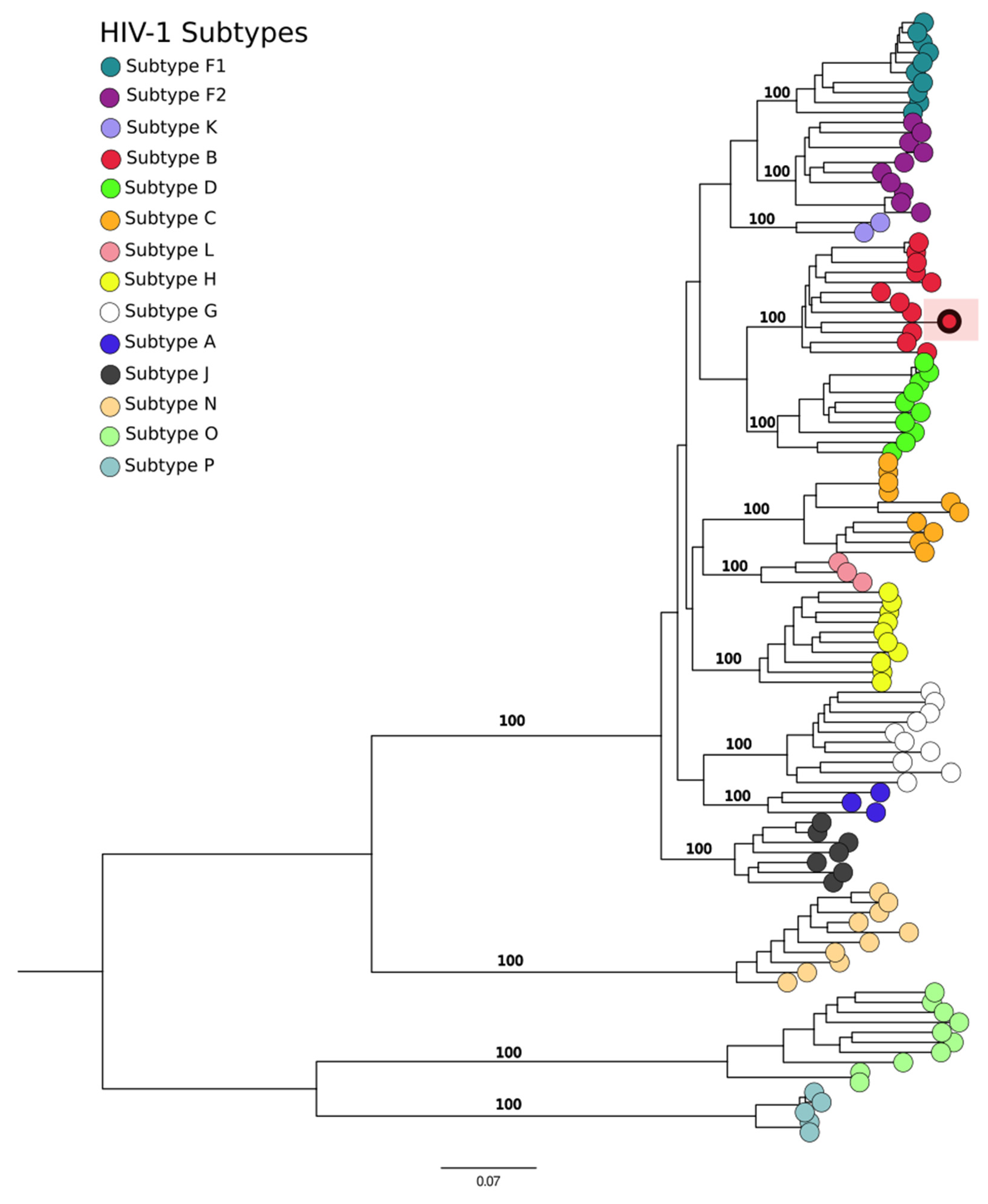
2.4. Phylogenetic Analysis
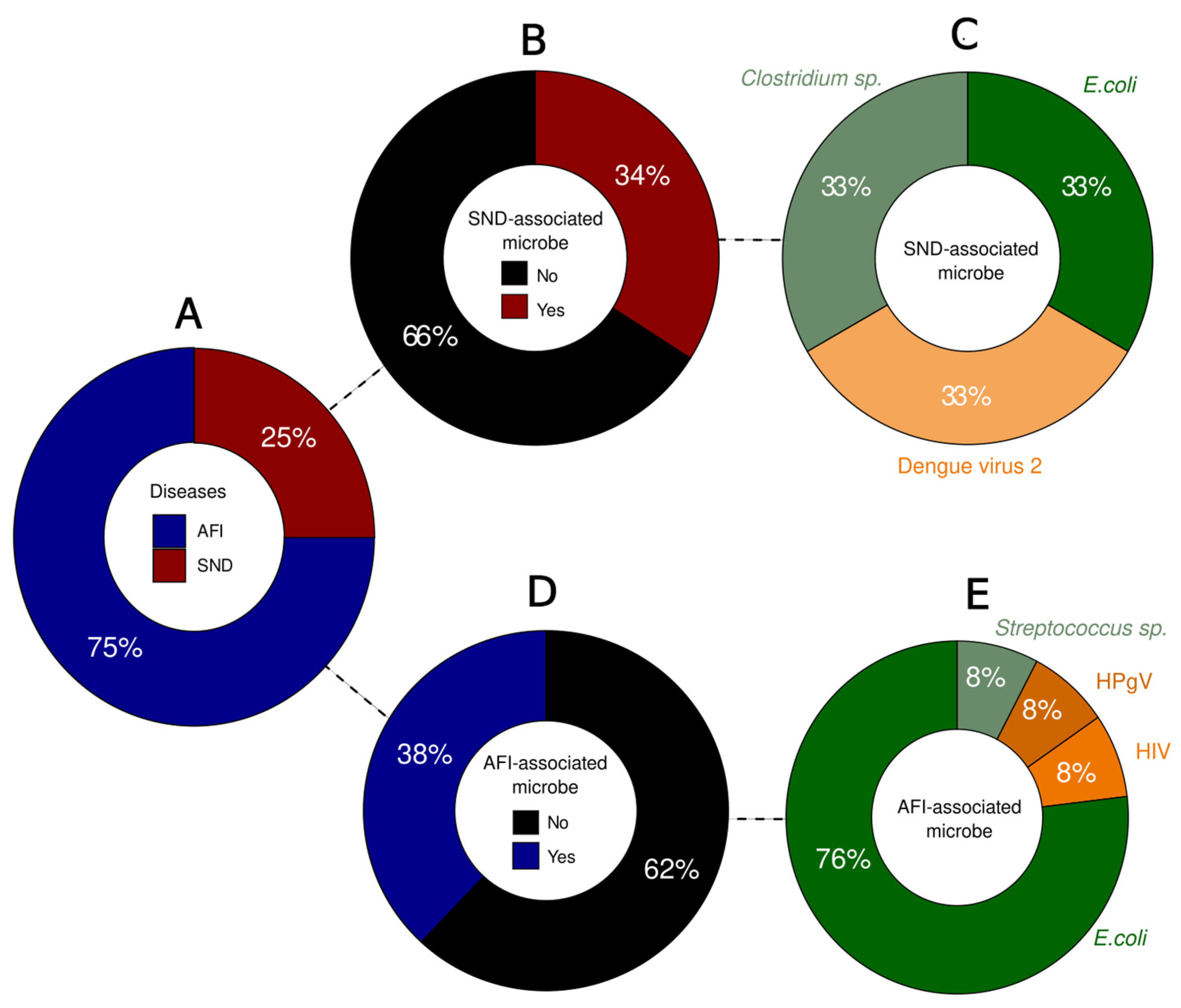
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mbidde, E.K.; Lutwama, J.J.; Perniciaro, J.L.; Nicholson, W.L.; Bower, W.A.; Bwogi, J.; Blaney, D.D. Investigating the etiology of acute febrile illness: A prospective clinic-based study in Uganda. BMC Infect. Dis. 2023, 23, 411. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tam, P.Y.I.; Obaro, S.K.; Storch, G. Challenges in the Etiology and Diagnosis of Acute Febrile Illness in Children in Low- and Middle-Income Countries. J. Pediatr. Infect. Dis. Soc. 2016, 5, 190–205. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moreira, J.; Bressan, C.S.; Brasil, P.; Siqueira, A.M. Epidemiology of acute febrile illness in Latin America. Clin. Microbiol. Infect. 2018, 24, 827–835. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bressan, C.D.S.; Teixeira, M.L.B.; Gouvêa, M.I.F.D.S.; de Pina-Costa, A.; Santos, H.F.P.; Calvet, G.A.; Lupi, O.; Siqueira, A.M.; Valls-de-Souza, R.; Valim, C.; et al. Challenges of acute febrile illness diagnosis in a national infectious diseases center in Rio de Janeiro: 16-year experience of syndromic surveillance. PLoS Negl. Trop. Dis. 2023, 17, e0011232. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, X.; Laurent, S.; Onur, O.A.; Kleineberg, N.N.; Fink, G.R.; Schweitzer, F.; Warnke, C. A systematic review of neurological symptoms and complications of COVID-19. J. Neurol. 2021, 268, 392–402. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wouk, J.; Rechenchoski, D.Z.; Rodrigues, B.C.D.; Ribelato, E.V.; Faccin-Galhardi, L.C. Viral infections and their relationship to neurological disorders. Arch. Virol. 2021, 166, 733–753. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ciuffreda, L.; Rodríguez-Pérez, H.; Flores, C. Nanopore sequencing and its application to the study of microbial communities. Comput. Struct. Biotechnol. J. 2021, 19, 1497–1511. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Deng, Q.; Cao, Y.; Wan, X.; Wang, B.; Sun, A.; Wang, H.; Wang, Y.; Wang, H.; Gu, H. Nanopore-based metagenomic sequencing for the rapid and precise detection of pathogens among immunocompromised cancer patients with suspected infections. Front. Cell Infect. Microbiol. 2022, 12, 943859. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Latorre-Pérez, A.; Villalba-Bermell, P.; Pascual, J.; Vilanova, C. Assembly methods for nanopore-based metagenomic sequencing: A comparative study. Sci. Rep. 2020, 10, 13588. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Claro, I.M.; Ramundo, M.S.; Coletti, T.M.; da Silva, C.A.M.; Valenca, I.N.; Candido, D.S.; Sales, F.C.S.; Manuli, E.R.; de Jesus, J.G.; de Paula, A.; et al. Rapid viral metagenomics using SMART-9N amplification and nanopore sequencing. Wellcome Open Res. 2023, 6, 241. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kalantar, K.L.; Carvalho, T.; de Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.F.; King, R.; et al. IDseq-An open-source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. GigaScience 2020, 9, giaa111. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P.L.; et al. metaFlye: Scalable long-read metagenome assembly using repeat graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.M.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534, Erratum in Mol. Biol. Evol. 2020, 37, 2461. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rambaut, A. FigTree v1.3.1. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh. 2010. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 17 June 2024).
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Schuppner, R.; Maehlmann, J.; Dirks, M.; Worthmann, H.; Tryc, A.B.; Sandorski, K.; Bahlmann, E.; Kielstein, J.T.; Giesemann, A.M.; Lanfermann, H.; et al. Neurological Sequelae in Adults After E coli O104: H4 Infection-Induced Hemolytic-Uremic Syndrome. Medicine 2016, 95, e2337. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Soedarmono, P.; Diana, A.; Tauran, P.; Lokida, D.; Aman, A.T.; Alisjahbana, B.; Arlinda, D.; Tjitra, E.; Kosasih, H.; Merati, K.T.P.; et al. The characteristics of bacteremia among patients with acute febrile illness requiring hospitalization in Indonesia. PLoS ONE 2022, 17, e0273414. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Daga, A.P.; Koga, V.L.; Soncini, J.G.M.; de Matos, C.M.; Perugini, M.R.E.; Pelisson, M.; Kobayashi, R.K.T.; Vespero, E.C. Escherichia coli Bloodstream Infections in Patients at a University Hospital: Virulence Factors and Clinical Characteristics. Front. Cell Infect. Microbiol. 2019, 9, 191. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Junqueira, D.M.; Almeida, S.E. HIV-1 subtype B: Traces of a pandemic. Virology 2016, 495, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, G.J.; Aleixo, A.W.; Tupinambás, U. Prevalence of HIV-1 transmitted drug resistance and its impact on the effectiveness of antiretroviral therapy—Minas Gerais state, Brazil. Braz. J. Health Rev. 2022, 5, 1044–1059. [Google Scholar] [CrossRef]
- Yu, Y.; Wan, Z.; Wang, J.H.; Yang, X.; Zhang, C. Review of human pegivirus: Prevalence, transmission, pathogenesis, and clinical implication. Virulence 2022, 13, 324–341. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Finsterer, J.; Hess, B. Neuromuscular and central nervous system manifestations of Clostridium perfringens infections. Infection 2007, 35, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Kumar, R.; Singh, B.R. Clostridial Neurotoxins: Structure, Function and Implications to Other Bacterial Toxins. Microorganisms 2021, 9, 2206. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Trivedi, S.; Chakravarty, A. Neurological Complications of Dengue Fever. Curr. Neurol. Neurosci. Rep. 2022, 22, 515–529. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kulkarni, R.; Pujari, S.; Gupta, D. Neurological Manifestations of Dengue Fever. Ann. Indian. Acad. Neurol. 2021, 24, 693–702. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Giovanetti, M.; Pereira, L.A.; Santiago, G.A.; Fonseca, V.; Mendoza, M.P.G.; de Oliveira, C.; de Moraes, L.; Xavier, J.; Tosta, S.; Fristch, H.; et al. Emergence of Dengue Virus Serotype 2 Cosmopolitan Genotype, Brazil. Emerg. Infect. Dis. 2022, 2, 1725–1727. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Amorim, M.T.; Hernández, L.H.A.; Naveca, F.G.; Essashika Prazeres, I.T.; Wanzeller, A.L.M.; Silva, E.V.P.D.; Casseb, L.M.N.; Silva, F.S.D.; da Silva, S.P.; Nunes, B.T.D.; et al. Emergence of a New Strain of DENV-2 in South America: Introduction of the Cosmopolitan Genotype through the Brazilian-Peruvian Border. Trop. Med. Infect. Dis. 2023, 8, 325. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Arboviruses Epidemic. Available online: https://www.saude.mg.gov.br/component/gmg/story/18774-epidemia-arboviroses (accessed on 12 April 2024).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gender | Age | City | State | Clinical Classification | Symptoms | Sample |
|---|---|---|---|---|---|---|---|
| 1 | Male | 53 | Belo Horizonte | Minas Gerais | SND | Headaches | Serum |
| 2 | Female | 38 | Belo Horizonte | Minas Gerais | AFI | Fever, myalgia, arthralgia, fatigue, headache, and skin lesions (rash) | Tempus |
| 3 | Male | 38 | Belo Horizonte | Minas Gerais | SND | Headache, convulsive seizure, and neurological symptoms | Whole blood |
| 4 | Male | 53 | Ouro Preto | Minas Gerais | SND | Suggestive of neurosyphilis and bacterial meningitis | Whole blood |
| 5 | Female | 43 | Belo Horizonte | Minas Gerais | AFI | Myalgia, arthralgia, fatigue, and petechiae | Serum |
| 6 | Male | 34 | Belo Horizonte | Minas Gerais | AFI | Fever, myalgia, headache, and petechiae | Whole blood |
| 7 | Female | 27 | Ribeirão das Neves | Minas Gerais | AFI | Fever, cough, myalgia, fatigue, and petechiae | Tempus, serum |
| 8 | Female | 14 | Belo Horizonte | Minas Gerais | AFI | Fever, myalgia, and headache | Whole blood |
| 9 | Male | 37 | Belo Horizonte | Minas Gerais | AFI | Fever | Serum |
| 10 | Male | 61 | Betim | Minas Gerais | AFI | Fever | Serum |
| 11 | Female | 23 | Belo Horizonte | Minas Gerais | AFI | Fever and subacute myeloradiculopathy | Cerebrospinal fluids |
| 12 | Female | 32 | Belo Horizonte | Minas Gerais | AFI | Fever, myalgia, skin lesions, immunosuppression, and acute hepatitis | Whole blood |
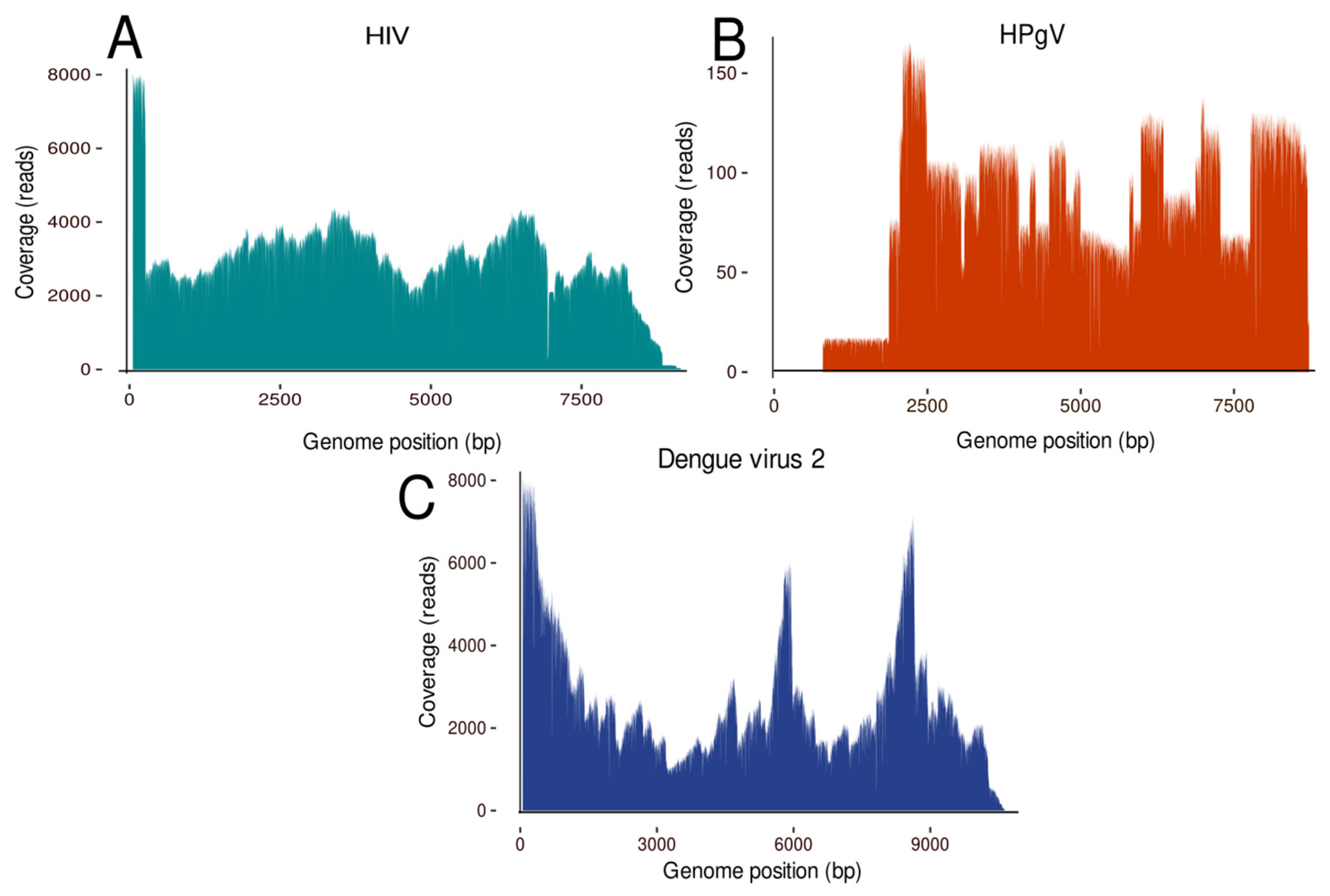
| Patient | Sample Type | Pathogen | Reads | Contigs | Coverage | Depth |
|---|---|---|---|---|---|---|
| 1 | Serum | Clostridium sp. | 6554 | 1 | 0.8% | 0.25× |
| 2 | Tempus | E. coli | 63 | 1 | 0.1% | 0.005× |
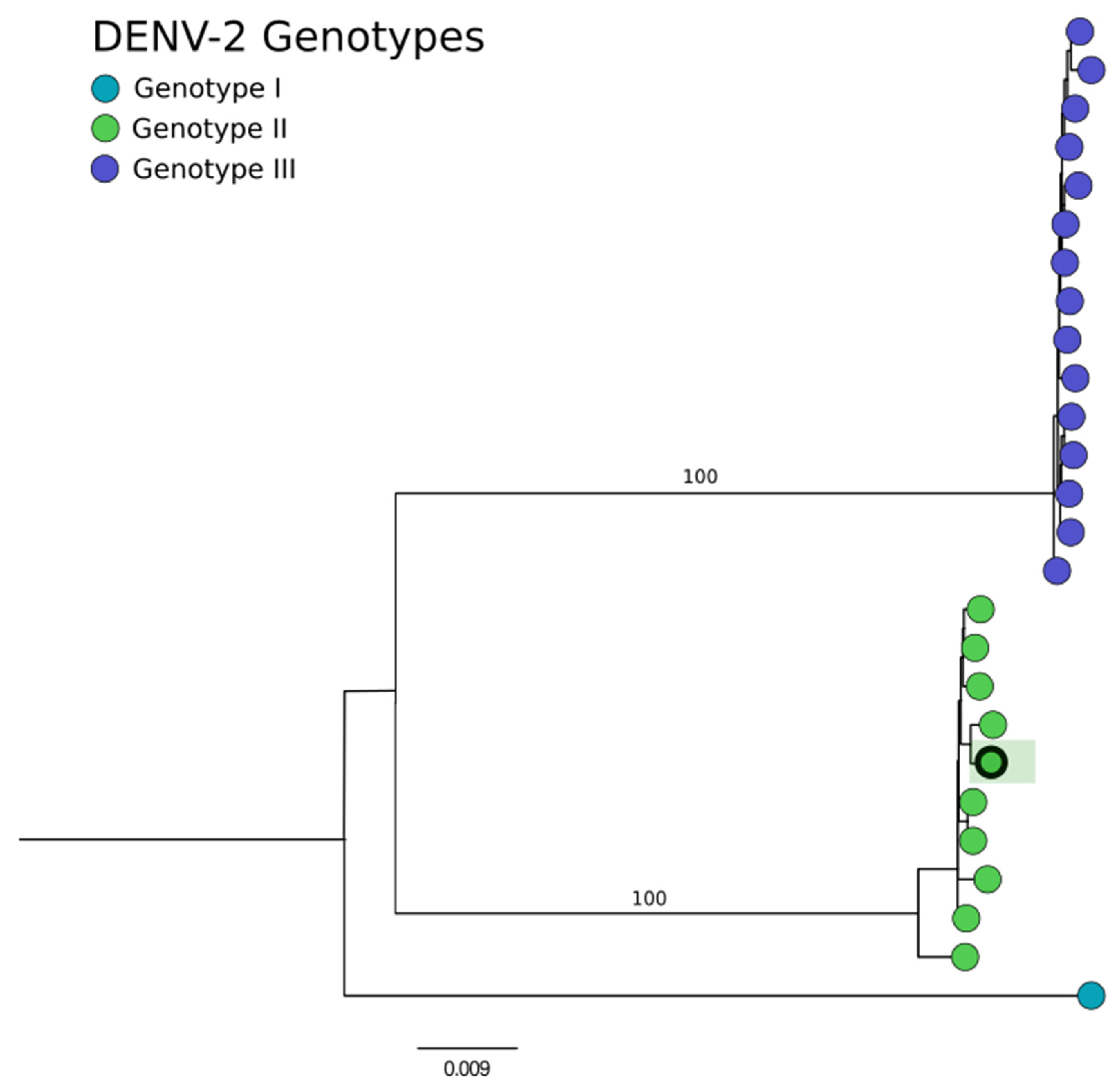
| 3 | Whole blood | DENV-2 | 72,078 | 3 | 99.8% | 1145× |
| 4 | Whole blood | E. coli | 6939 | 1 | 0.1% | 0.61× |
| 5 | Serum | E. coli | 10,968 | 1 | 0.3% | 1× |
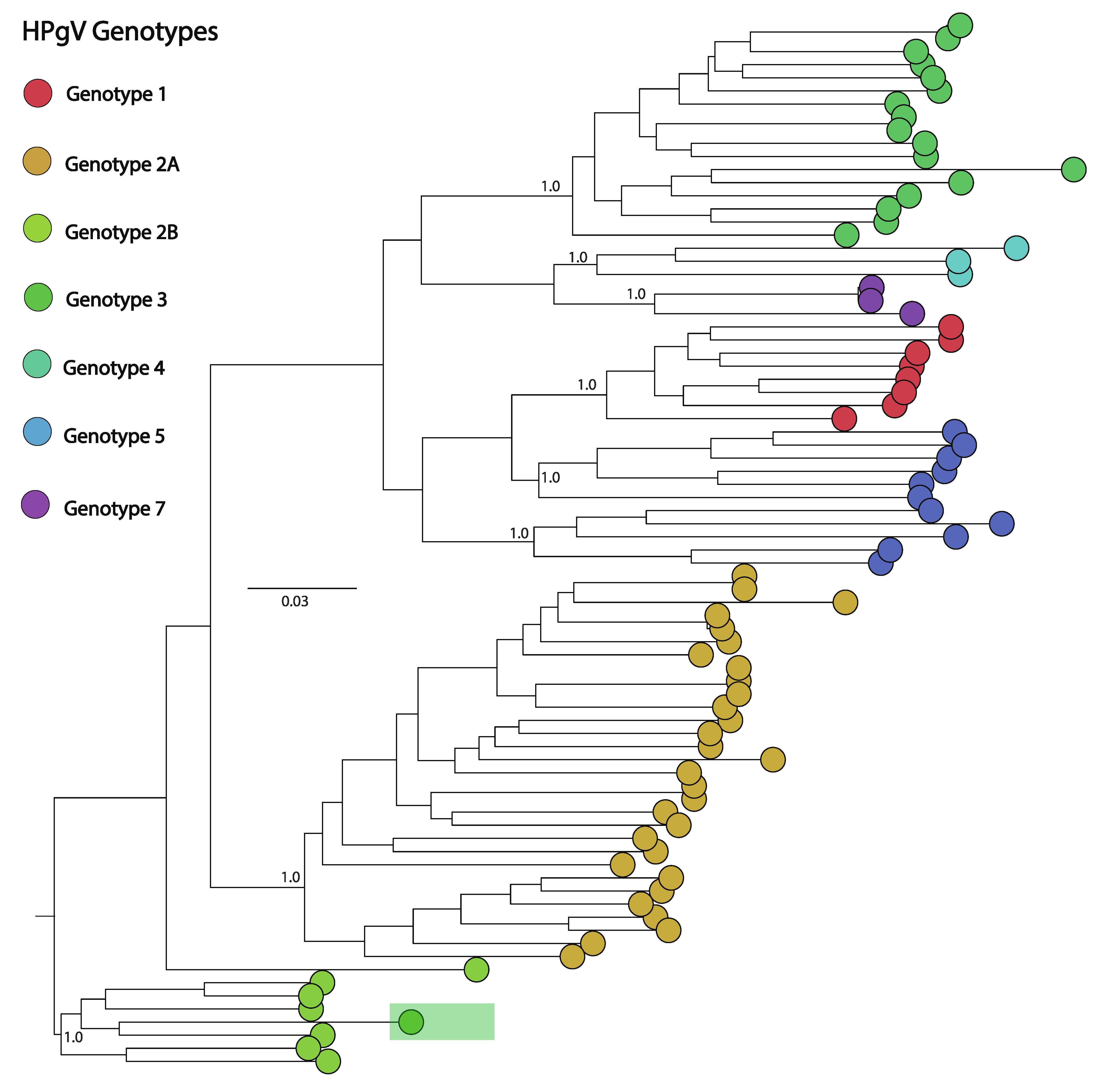
| 6 | Whole blood | HPgV | 511 | 2 | 86.2% | 41× |
| E. coli | 4635 | 1 | 0.4% | 0.18× | ||
| 7 | Streptococcus sp. | 1347 | 1 | 0.1% | 0.17× | |
| Tempus | E. coli | 173 | 1 | 0.7% | 0.012× | |
| Serum | E. coli | 9889 | 1 | 0.1% | 0.95× | |
| 8 | Whole blood | E. coli | 5280 | 1 | 0.1% | 0.41× |
| 9 | Serum | HIV-1 | 52,420 | 2 | 92% | 1805× |
| E. coli | 13,778 | 1 | 0.2% | 1.2× | ||
| 10 | Serum | E. coli | 22,622 | 1 | 0.1% | 1.6× |
| 11 | Cerebrospinal fluid 1 | E. coli | 2084 | 1 | 0.1% | 0.19× |
| Cerebrospinal fluid 2 | E. coli | 1703 | 1 | 0.2% | 0.11× | |
| 12 | Whole blood | - | - | - | - | - |
| 13 | Negative control | E. coli | 27 | 0 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno, K.M.F.; de Andrade, V.A.; de Melo Iani, F.C.; Fonseca, V.; Lima, M.T.; de Castro Barbosa, E.; Tomé, L.M.R.; Guimarães, N.R.; Fritsch, H.M.; Adelino, T.; et al. Exploring Microorganisms Associated to Acute Febrile Illness and Severe Neurological Disorders of Unknown Origin: A Nanopore Metagenomics Approach. Genes 2024, 15, 922. https://doi.org/10.3390/genes15070922
Moreno KMF, de Andrade VA, de Melo Iani FC, Fonseca V, Lima MT, de Castro Barbosa E, Tomé LMR, Guimarães NR, Fritsch HM, Adelino T, et al. Exploring Microorganisms Associated to Acute Febrile Illness and Severe Neurological Disorders of Unknown Origin: A Nanopore Metagenomics Approach. Genes. 2024; 15(7):922. https://doi.org/10.3390/genes15070922
Chicago/Turabian StyleMoreno, Keldenn Melo Farias, Virgínia Antunes de Andrade, Felipe Campos de Melo Iani, Vagner Fonseca, Maurício Teixeira Lima, Emerson de Castro Barbosa, Luiz Marcelo Ribeiro Tomé, Natália Rocha Guimarães, Hegger Machado Fritsch, Talita Adelino, and et al. 2024. "Exploring Microorganisms Associated to Acute Febrile Illness and Severe Neurological Disorders of Unknown Origin: A Nanopore Metagenomics Approach" Genes 15, no. 7: 922. https://doi.org/10.3390/genes15070922