
A Comparison of Structural Variant Calling from Short-Read and Nanopore-Based Whole-Genome Sequencing Using Optical Genome Mapping as a Benchmark

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics
2.2. Genome Sequencing
2.3. Whole-Genome Sequencing Using Oxford Nanopore Technologies (ONT)
2.4. Optical Genome Mapping (OGM) Using the Bionano Platform
3. Results
3.1. ONT and OGM Run Qualities
3.2. Bionano Benchmarks and Reference SV Set
3.3. Illumina and ONT WGS Performance Evaluation
3.4. ONT Performance—De Novo Calling
4. Discussion
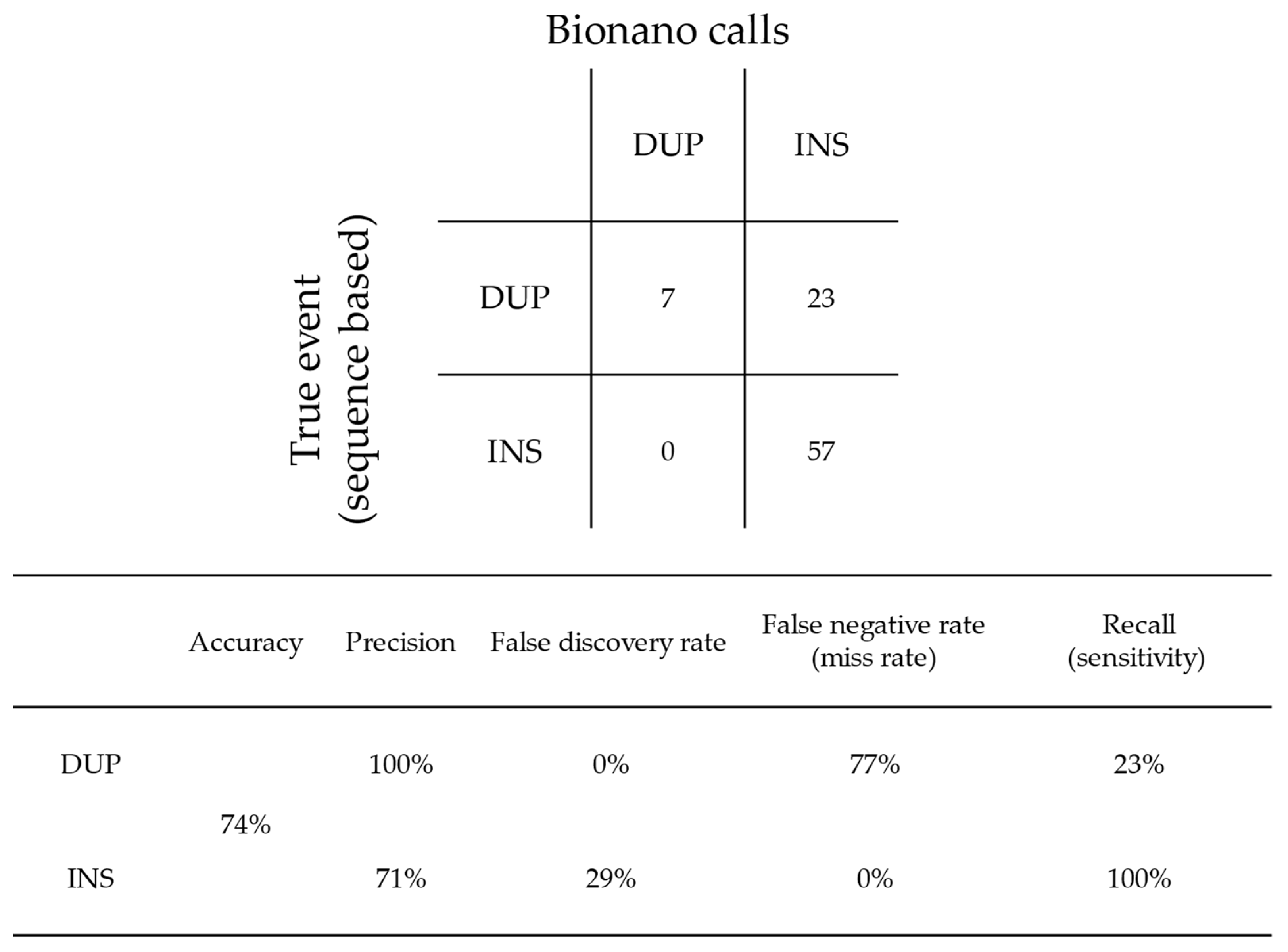
- Bionano OGM is highly precise, with 95% of rare SVs called as true events verified by molecules and/or reads supported from at least two technologies. However, Bionano OGM also misclassified 77% of tandem DUP as INS, owing to insufficient resolution for smaller DUPs, usually less than ~10 kb.
- Illumina WGS, with the union of three SV callers, successfully called 71% of events from the Bionano reference callset, with the most poorly detected SV type being INS at only 22% sensitivity.
- ONT performance is highly dependent on the variant caller. The original Sniffles performed poorly with a 48% sensitivity, and Sniffles2 demonstrated a significant improvement with 84% sensitivity.
- ONT performs much better at INS detection compared to Illumina.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bagger, F.O.; Borgwardt, L.; Jespersen, A.S.; Hansen, A.R.; Bertelsen, B.; Kodama, M.; Nielsen, F.C. Whole genome sequencing in clinical practice. BMC Med. Genom. 2024, 17, 39. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Chowdhury, S.; Taft, R.J.; Lebo, M.S.; Buchan, J.G.; Harrison, S.M.; Rowsey, R.; Klee, E.W.; Liu, P.F.; Worthey, E.A.; et al. Best practices for the analytical validation of clinical whole-genome sequencing intended for the diagnosis of germline disease. NPJ Genom. Med. 2020, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Austin-Tse, C.A.; Jobanputra, V.; Perry, D.L.; Bick, D.; Taft, R.J.; Venner, E.; Gibbs, R.A.; Young, T.; Barnett, S.; Belmont, J.W.; et al. Best practices for the interpretation and reporting of clinical whole genome sequencing. NPJ Genom. Med. 2022, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Stranneheim, H.; Lagerstedt-Robinson, K.; Magnusson, M.; Kvarnung, M.; Nilsson, D.; Lesko, N.; Engvall, M.; Anderlid, B.M.; Arnell, H.; Johansson, C.B.; et al. Integration of whole genome sequencing into a healthcare setting: High diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 2021, 13, 40. [Google Scholar] [CrossRef]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Turro, E.; Astle, W.J.; Megy, K.; Gräf, S.; Greene, D.; Shamardina, O.; Allen, H.L.; Sanchis-Juan, A.; Frontini, M.; Thys, C.; et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature 2020, 583, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Hyder, Z.; Calpena, E.; Pei, Y.; Tooze, R.S.; Brittain, H.; Twigg, S.R.F.; Cilliers, D.; Morton, J.E.V.; McCann, E.; Weber, A.; et al. Evaluating the performance of a clinical genome sequencing program for diagnosis of rare genetic disease, seen through the lens of craniosynostosis. Genet. Med. 2021, 23, 2360–2368. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Marmiesse, A.; Gouveia, S.; Couce, M.L. NGS Technologies as a Turning Point in Rare Disease Resea rch, Diagnosis and Treatment. Curr. Med. Chem. 2018, 25, 404–432. [Google Scholar] [CrossRef]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.Y.; et al. An integrated map of structural variation in 2504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef]
- Currall, B.B.; Chiangmai, C.; Talkowski, M.E.; Morton, C.C. Mechanisms for Structural Variation in the Human Genome. Curr. Genet. Med. Rep. 2013, 1, 81–90. [Google Scholar] [CrossRef]
- Pinto, D.; Marshall, C.; Feuk, L.; Scherer, S.W. Copy-number variation in control population cohorts. Hum. Mol. Genet. 2007, 16, R168–R173. [Google Scholar] [CrossRef] [PubMed]
- Simon-Sanchez, J.; Scholz, S.; Fung, H.C.; Matarin, M.; Hernandez, D.; Gibbs, J.R.; Britton, A.; de Vrieze, F.W.; Peckham, E.; Gwinn-Hardy, K.; et al. Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum. Mol. Genet. 2007, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shira, A.; Rosner, G.; Rosner, S.; Goldstein, M.; Orr-Urtreger, A. Array-based comparative genome hybridization in clinical genetics. Pediatr. Res. 2006, 60, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Mandujano, G.; Grünberg, R.; Zhang, Y.; Bi, C.; Guzmán-Vega, F.J.; Shuaib, M.; Gorchakov, R.V.; Xu, J.; Tehseen, M.; Takahashi, M.; et al. An open-source, automated, and cost-effective platform for COVID-19 diagnosis and rapid portable genomic surveillance using nanopore sequencing. Sci. Rep. 2023, 13, 20349. [Google Scholar] [CrossRef] [PubMed]
- Pang, A. Comprehensive detection of germline and somatic structural mutation in cancer genomes by Bionano Genomics optical mapping. Cancer Res. 2019, 79, 5122. [Google Scholar] [CrossRef]
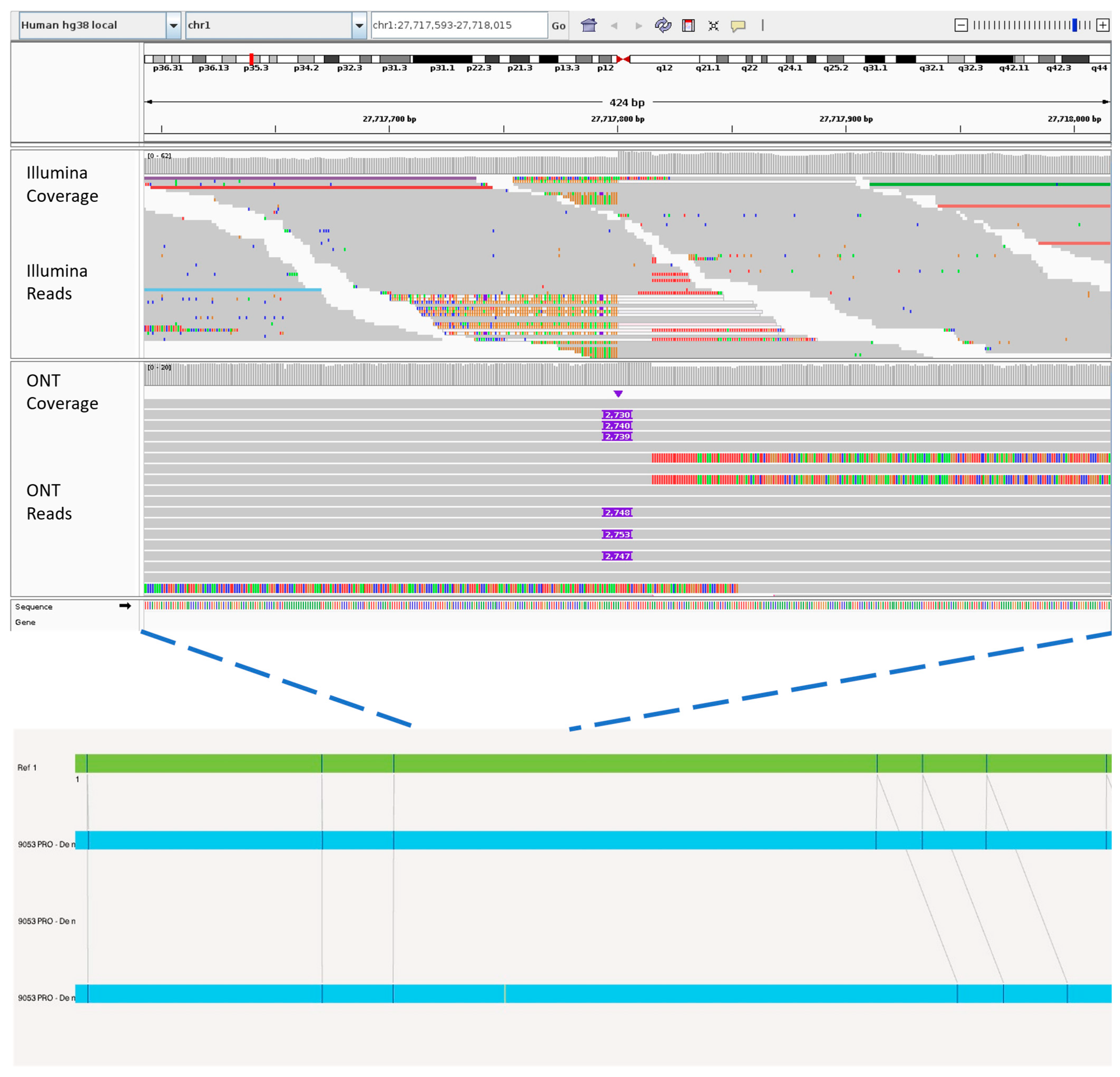
- Bjornstad, P.M.; Aalokken, R.; Åsheim, J.; Sundaram, A.Y.M.; Felde, C.N.; Ostby, G.H.; Dalland, M.; Sjursen, W.; Carrizosa, C.; Vigeland, M.D.; et al. A 39 kb structural variant causing Lynch Syndrome detected by optical genome mapping and nanopore sequencing. Eur. J. Hum. Genet. 2023, 32, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S.; Matsumoto, N. Long-read sequencing for rare human genetic diseases. J. Hum. Genet. 2020, 65, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, A.O.M.; Johnson, D.; Wall, S.A. Clinical genetics of craniosynostosis. Curr. Opin. Pediatr. 2017, 29, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Smedley, D.; Smith, K.R.; Martin, A.; Thomas, E.A.; McDonagh, E.M.; Cipriani, V.; Ellingford, J.M.; Arno, G.; Tucci, A.; Vandrovcova, J.; et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care—Preliminary Report. N. Engl. J. Med. 2021, 385, 1868–1880. [Google Scholar] [CrossRef]
- Caulfield, M.; Davies, J.; Dennys, M.; Elbahy, L.; Fowler, T.; Hill, S.; Hubbard, T.; Jostins, L.; Maltby, N.; Mahon-Pearson, J.; et al. National Genomic Research Library. 2020. Available online: https://doi.org/10.6084/m9.figshare.4530893.v7 (accessed on 15 July 2024).
- Chen, X.Y.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Kallberg, M.; Cox, A.J.; Kruglyakl, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Roller, E.; Ivakhno, S.; Lee, S.; Royce, T.; Tanner, S. Canvas: Versatile and scalable detection of copy number variants. Bioinformatics 2016, 32, 2375–2377. [Google Scholar] [CrossRef] [PubMed]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y. Characterising Structural Variants in Patients with Craniosynostosis Using Short-Read and Long-Range Technologies. Ph.D. Thesis, University of Oxford, Oxford, UK, 2024. Available online: https://doi.org/10.5287/ora-ovqzxnzr2 (accessed on 10 July 2024).
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, E31–E34. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.; Paulin, L.; Grochowski, C.; Horner, D.; Mahmoud, M.; Behera, S.; Kalef-Ezra, E.; Gandhi, M.; Hong, K.; Pehlivan, D.; et al. Detection of mosaic and population-level structural variants with Sniffles2. Nat. Biotechnol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Savara, J.; Novosád, T.; Gajdos, P.; Kriegová, E. Comparison of structural variants detected by optical mapping with long-read next-generation sequencing. Bioinformatics 2021, 37, 3398–3404. [Google Scholar] [CrossRef] [PubMed]
- Talsania, K.; Shen, T.; Chen, X.; Jaeger, E.; Li, Z.; Chen, Z.; Chen, W.; Tran, B.; Kusko, R.; Wang, L.; et al. Structural variant analysis of a cancer reference cell line sample using multiple sequencing technologies. Genome Biol. 2022, 23, 255. [Google Scholar] [CrossRef]
- Du, X.; Li, L.; Liang, F.; Liu, S.; Zhang, W.; Sun, S.; Sun, Y.; Fan, F.; Wang, L.; Liang, X.; et al. Robust Benchmark Structural Variant Calls of An Asian Using State-of-the-art Long-read Sequencing Technologies. Genom. Proteom. Bioinform. 2022, 20, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Zook, J.M.; Hansen, N.F.; Olson, N.D.; Chapman, L.; Mullikin, J.C.; Xiao, C.N.; Sherry, S.; Koren, S.; Phillippy, A.M.; Boutros, P.C.; et al. A robust benchmark for detection of germline large deletions and insertions. Nat. Biotechnol. 2020, 38, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, S.; Momozawa, Y.; Liu, X.X.; Terao, C.; Kubo, M.; Kamatani, Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019, 20, 117. [Google Scholar] [CrossRef]
- Dremsek, P.; Schwarz, T.; Weil, B.; Malashka, A.; Laccone, F.; Neesen, J. Optical Genome Mapping in Routine Human Genetic Diagnostics-Its Advantages and Limitations. Genes 2021, 12, 1958. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Werling, D.M.; Brand, H.; An, J.Y.; Stone, M.R.; Zhu, L.X.; Glessner, J.T.; Collins, R.L.; Dong, S.; Layer, R.M.; Markenscoff-Papadimitriou, E.; et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 2018, 50, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alfoldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| BATCH 1 | BATCH 2 | BATCH 3 | |
|---|---|---|---|
| Number of proband samples | 2 | 4 | 3 |
| ONT Flow Cell | R9.4 | R9.4 | R10 |
| Guppy | 5.0.12 (on sequencer) | 5.0.7 (on server) | 6.4.6 (on server) |
| Minimap2 a | 2.20-r1061 | 2.24-r1122 | 2.24-r1122 |
| Sniffles a | 1.0.11 | 1.0.11 | 2.0.6 |
| Samtools a | 1.11 | 1.11 | 1.11 |
| Tabix a | 1.9 | 1.9 | 1.9 |
| ONT Metrics | OGM Metrics | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fam ID | Case ID | Sample | Batch | Estimated Bases Produced (Gb) | Estimated N50 (kb) | Estimated Coverage (×) | Total DNA (≥20 kb), Gb a | Total DNA (≥150 kb), Gb a | N50 (≥20 kb), kb b | N50 (≥150 kb), kb b | Average Label Density (≥150 kb),/100 kb c | Map Rate (≥150 kb) d | Effective Coverage (×) e | Molecule Integrity Number f | Positive Label Variance (PLV) g | Negative Label Variance (NLV) h |
| crs510 | 1 | Proband | 1 | 84.33 | 29.38 | 26 | 675.6 | 507.8 | 265.5 | 328.9 | 14.3 | 84.1% | 132.08 | 0.08 | 2.2% | 14.6% |
| Mother | 1 | 95.94 | 24.76 | 30 | ||||||||||||
| Father | 1 | 90.91 | 27.75 | 28 | ||||||||||||
| L16 | 2 | Proband | 1 | 156.92 | 26.62 | 49 | 944.3 | 768.1 | 336.4 | 409.9 | 17.9 | 81.5% | 195.79 | 0.09 | 2.6% | 9.0% |
| Mother | 1 | 104.41 | 24.59 | 33 | ||||||||||||
| Father | 1 | 115.23 | 28 | 36 | ||||||||||||
| M196 | 3 | Proband | 2 | 9.94 | 27.72 | 3 | 965.9 | 510.7 | 159.8 | 267.4 | 15.3 | 51.2% | 77.88 | 0.12 | 3.0% | 18.6% |
| Mother | 2 | 13.31 | 22.26 | 4 | ||||||||||||
| Father | 2 | 37.93 | 19.73 | 12 | ||||||||||||
| crs169 | 4 | Proband | 2 | 48.88 | 22.6 | 15 | 3189.4 | 1527.1 | 125.6 | 409.5 | 16.4 | 90.8% | 432.94 | 0.07 | 2.9% | 6.4% |
| Mother | 2 | 46.88 | 18.96 | 15 | ||||||||||||
| Father | 2 | 55.98 | 34.31 | 17 | ||||||||||||
| crs290 | 5 | Proband | 2 | 56.59 | 30.91 | 18 | 2163 | 1529 | 239.2 | 317.3 | 16.1 | 89.9% | 417.7 | 0.1 | 2.4% | 7.7% |
| Mother | 2 | 73.86 | 18.73 | 23 | ||||||||||||
| Father | 2 | 47.56 | 24.24 | 15 | ||||||||||||
| crs685 | 6 | Proband | 2 | 85.31 | 29.92 | 27 | 681.3 | 542 | 272.6 | 320.2 | 15.8 | 93.4% | 157.1 | 0.08 | 2.4% | 7.2% |
| Mother | 2 | 92.52 | 28.3 | 29 | ||||||||||||
| Father | 2 | 82.81 | 35.21 | 26 | ||||||||||||
| crs566 | 7 | Proband | 3 | 95.96 | 21.89 | 30 | 752.8 | 604.6 | 302.6 | 356.3 | 15.4 | 92.7% | 173.54 | 0.07 | 4.3% | 6.0% |
| Mother | 2 | 49.94 | 30.66 | 16 | ||||||||||||
| Father | 2 | 35.71 | 32.48 | 11 | ||||||||||||
| crs302 | 8 | Proband | 3 | 73.77 | 19.24 | 23 | 1855.6 | 1525.6 | 290.6 | 333.5 | 15.6 | 94.7% | 450.36 | 0.07 | 2.3% | 6.8% |
| Mother | 3 | 85.13 | 18.69 | 27 | 2027.1 | 1504.4 | 253.2 | 315.7 | 15.0 | 91.9% | 430.46 | 0.07 | 2.1% | 10.8% | ||
| Father | 3 | 85.45 | 19.07 | 27 | 2091.7 | 1464.2 | 231.8 | 308.6 | 15.8 | 91.7% | 414.23 | 0.10 | 3.0% | 7.1% | ||
| M44 | 9 | Proband | 3 | 53.16 | 18.53 | 17 | 1960.1 | 1508.4 | 257.2 | 308.6 | 15.5 | 92.9% | 433.17 | 0.07 | 2.3% | 7.7% |
| Mother | 3 | 77.93 | 20.56 | 24 | ||||||||||||
| Father | 3 | 70.96 | 16.15 | 22 |
| CNV | CPX | DEL | Tandem DUP | INS | False/Uncertain | |
|---|---|---|---|---|---|---|
| OGM Calls | 3 | 1 | 136 | 12 | 82 | - |
| Reclassified/Normalised | 0 | 0 | 134 | 30 | 58 | 12 |
| Data Batches | Technology— SV Detection Method | Overall Performance | Performance by SV Type | |||||
|---|---|---|---|---|---|---|---|---|
| TP | FN | Sensitivity | SV Type | TP | FN | Sensitivity | ||
| Batch 1, 2, and 3 | Illumina—M + C + L a | DEL | 115 | 19 | 86% | |||
| 157 | 65 | 71% | DUP | 29 | 1 | 97% | ||
| INS | 13 | 45 | 22% | |||||
| Batch 1 and 2 R9 Flow Cells | ONT—Read Support b | DEL | 93 | 1 | 99% | |||
| 148 | 6 | 96% | DUP | 23 | 2 | 92% | ||
| INS | 32 | 3 | 91% | |||||
| ONT—Sniffles | DEL | 52 | 42 | 55% | ||||
| 74 | 80 | 48% | DUP | 9 | 16 | 36% | ||
| INS | 13 | 22 | 37% | |||||
| Batch 3 R10 Flow Cells | ONT—Read Support | DEL | 38 | 2 | 95% | |||
| 63 | 5 | 93% | DUP | 5 | 0 | 100% | ||
| INS | 20 | 3 | 87% | |||||
| ONT—Sniffles2 | DEL | 36 | 4 | 90% | ||||
| 57 | 11 | 84% | DUP | 4 | 1 | 80% | ||
| INS | 17 | 6 | 74% | |||||
| FP de novo a (Parental Call Missing) | TN de novo b (True Inherited Calls) | False Positive Rate (Probability of False de novo Calls) | ||
|---|---|---|---|---|
| Batches 1 and 2 | ONT–Sniffles | 19 | 55 | 26% |
| Batch 3 | ONT–Sniffles2 | 3 | 36 | 8% |
| Case | Chromosome | Position | SV Length | SV Type | Sniffles2-Ref Reads | Sniffles2-Alt Reads |
|---|---|---|---|---|---|---|
| 8 | chr15 | 43,572,861 | . | BND | 2 | 2 |
| 8 | chr19 | 1,993,025 | 70,128 | INS | 1 | 11 |
| 9 | chr12 | 52,517,168 | . | BND | 2 | 2 |
| 9 | chr16 | 69,961,666 | 6377 | INS | 5 | 3 |
| 9 | chr4 | 39,308,656 | . | BND | 4 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, Y.; Tanguy, M.; Giess, A.; Dixit, A.; Wilson, L.C.; Gibbons, R.J.; Twigg, S.R.F.; Elgar, G.; Wilkie, A.O.M. A Comparison of Structural Variant Calling from Short-Read and Nanopore-Based Whole-Genome Sequencing Using Optical Genome Mapping as a Benchmark. Genes 2024, 15, 925. https://doi.org/10.3390/genes15070925
Pei Y, Tanguy M, Giess A, Dixit A, Wilson LC, Gibbons RJ, Twigg SRF, Elgar G, Wilkie AOM. A Comparison of Structural Variant Calling from Short-Read and Nanopore-Based Whole-Genome Sequencing Using Optical Genome Mapping as a Benchmark. Genes. 2024; 15(7):925. https://doi.org/10.3390/genes15070925
Chicago/Turabian StylePei, Yang, Melanie Tanguy, Adam Giess, Abhijit Dixit, Louise C. Wilson, Richard J. Gibbons, Stephen R. F. Twigg, Greg Elgar, and Andrew O. M. Wilkie. 2024. "A Comparison of Structural Variant Calling from Short-Read and Nanopore-Based Whole-Genome Sequencing Using Optical Genome Mapping as a Benchmark" Genes 15, no. 7: 925. https://doi.org/10.3390/genes15070925
APA StylePei, Y., Tanguy, M., Giess, A., Dixit, A., Wilson, L. C., Gibbons, R. J., Twigg, S. R. F., Elgar, G., & Wilkie, A. O. M. (2024). A Comparison of Structural Variant Calling from Short-Read and Nanopore-Based Whole-Genome Sequencing Using Optical Genome Mapping as a Benchmark. Genes, 15(7), 925. https://doi.org/10.3390/genes15070925