
Replication of Known and Identification of Novel Associations in Biobank-Scale Datasets: A Survey Using UK Biobank and FinnGen

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Identification and Comparison of Associated Variants and Loci
2.3. Definition of Replicated and Non-Replicated Associations
3. Results
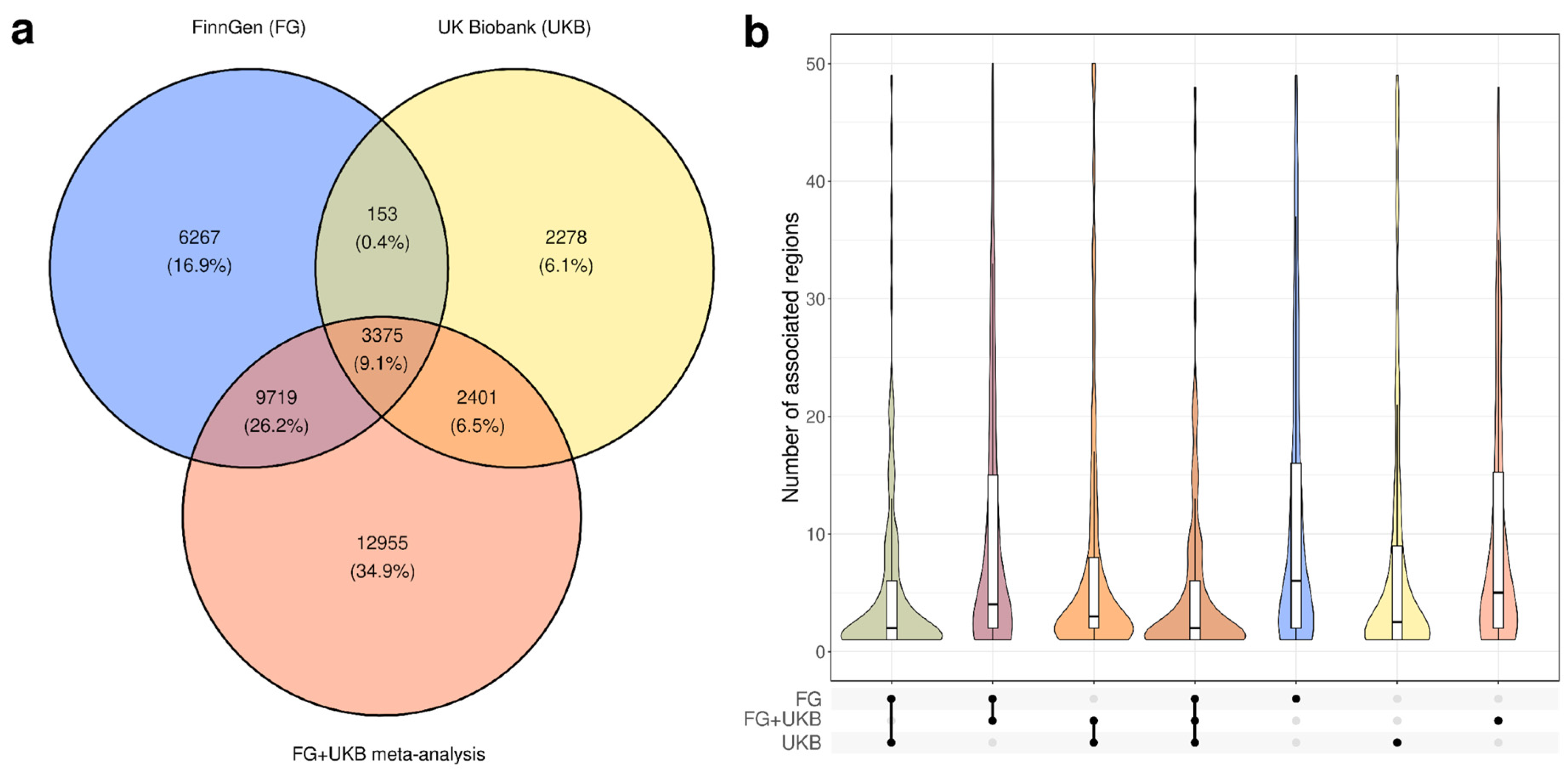
3.1. Characterization of Genome-Wide Significant Loci in UK Biobank and FinnGen
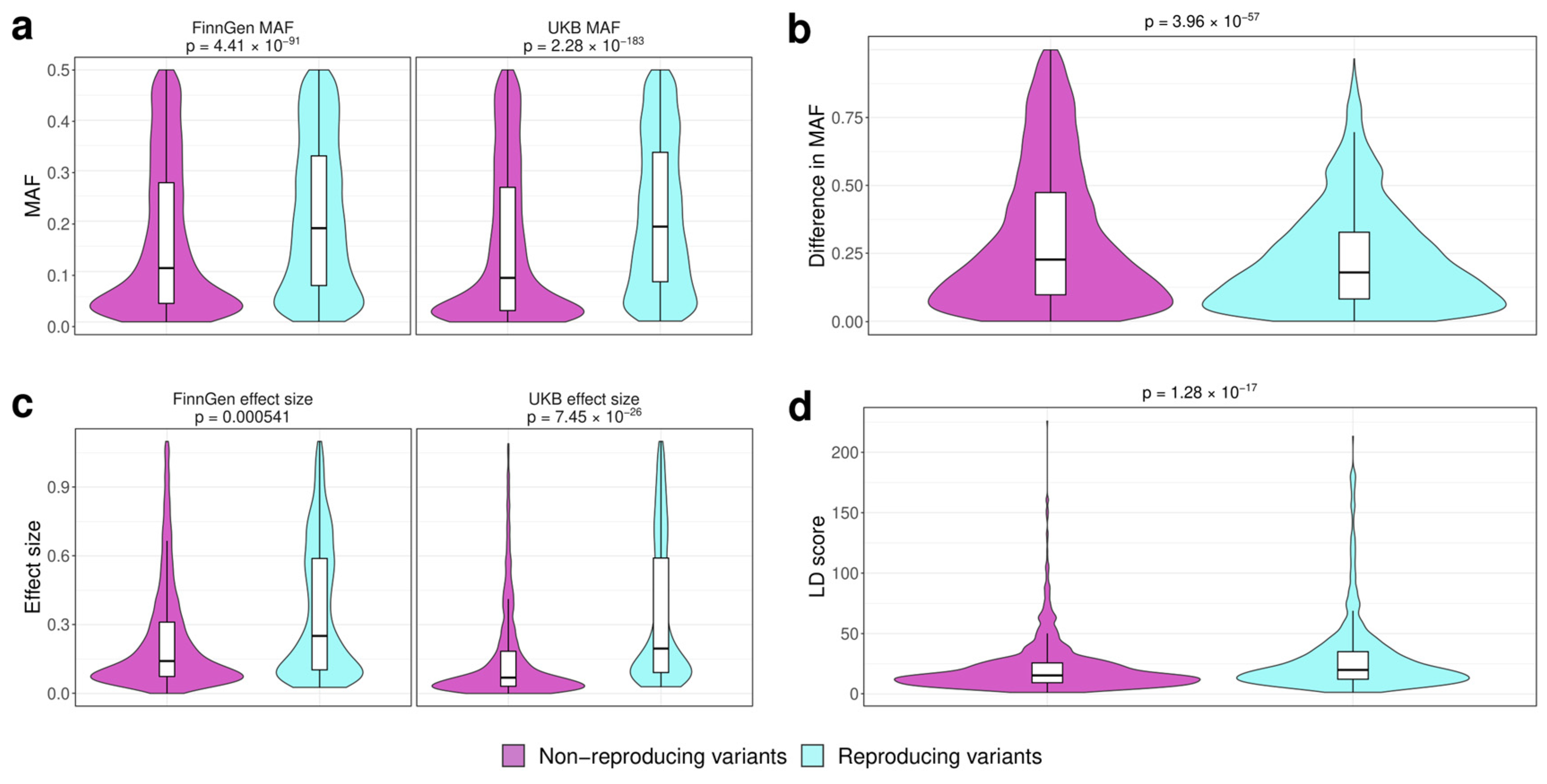
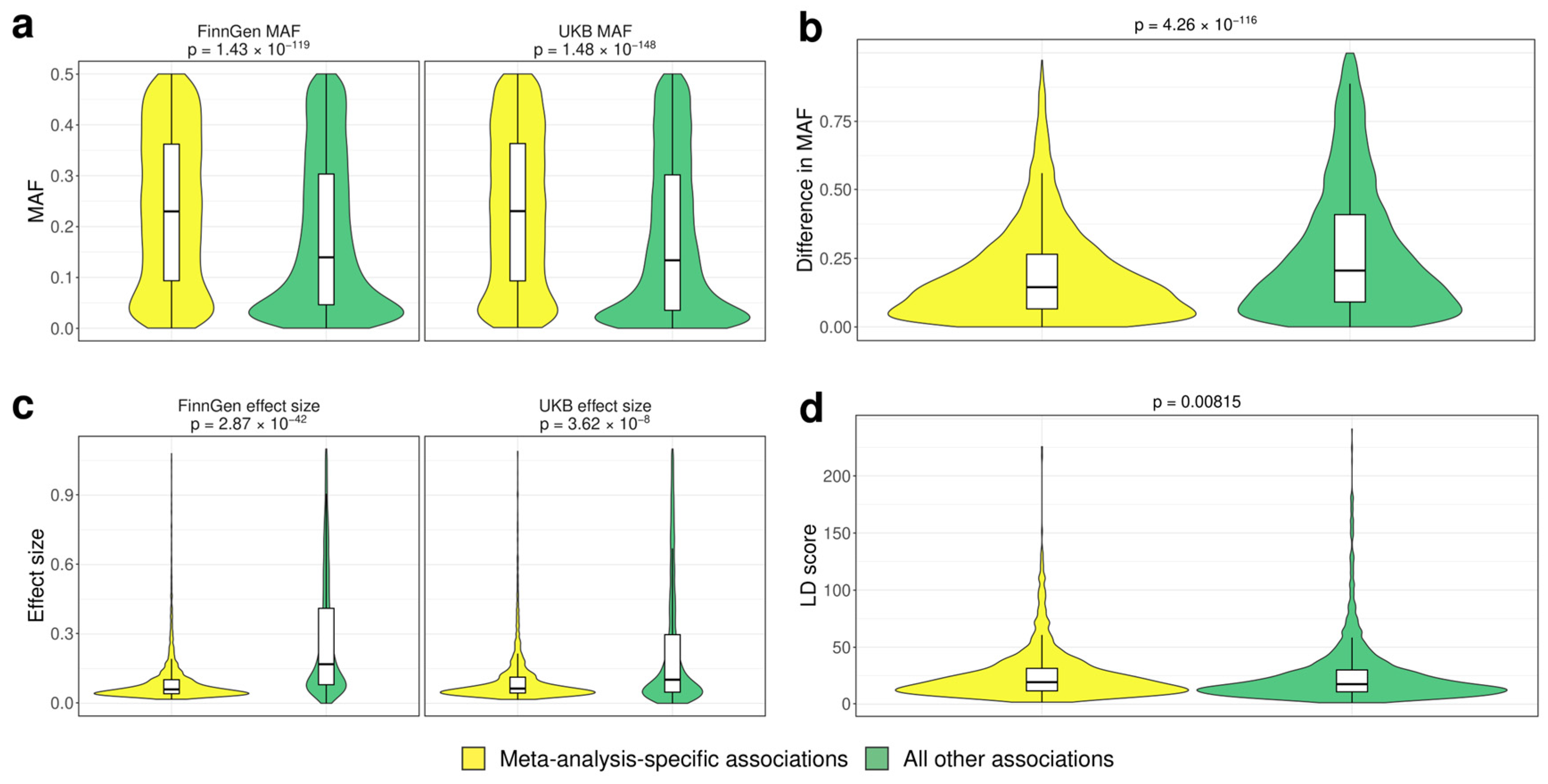
3.2. Analysis of the Factors Affecting Replication of Genome-Wide Associations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mills, M.C.; Rahal, C. A Scientometric Review of Genome-Wide Association Studies. Commun. Biol. 2019, 2, 9. [Google Scholar] [CrossRef]
- Loos, R.J.F. 15 Years of Genome-Wide Association Studies and No Signs of Slowing Down. Nat. Commun. 2020, 11, 5900. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of Published Genome-Wide Association Studies, Targeted Arrays and Summary Statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Marigorta, U.M.; Rodríguez, J.A.; Gibson, G.; Navarro, A. Replicability and Prediction: Lessons and Challenges from GWAS. Trends Genet. 2018, 34, 504–517. [Google Scholar] [CrossRef]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank Resource with Deep Phenotyping and Genomic Data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen Provides Genetic Insights from a Well-Phenotyped Isolated Population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef]
- Lazareva, T.E.; Barbitoff, Y.A.; Changalidis, A.I.; Tkachenko, A.A.; Maksiutenko, E.M.; Nasykhova, Y.A.; Glotov, A.S. Biobanking as a Tool for Genomic Research: From Allele Frequencies to Cross-Ancestry Association Studies. J. Pers. Med. 2022, 12, 2040. [Google Scholar] [CrossRef]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A Cross-Population Atlas of Genetic Associations for 220 Human Phenotypes. Nat. Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Hujoel, M.L.A.; Gazal, S.; Hormozdiari, F.; van de Geijn, B.; Price, A.L. Disease Heritability Enrichment of Regulatory Elements Is Concentrated in Elements with Ancient Sequence Age and Conserved Function across Species. Am. J. Hum. Genet. 2019, 104, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Okbay, A.; Beauchamp, J.P.; Fontana, M.A.; Lee, J.J.; Pers, T.H.; Rietveld, C.A.; Turley, P.; Chen, G.-B.; Emilsson, V.; Meddens, S.F.W.; et al. Genome-Wide Association Study Identifies 74 Loci Associated with Educational Attainment. Nature 2016, 533, 539–542. [Google Scholar] [CrossRef]
- Momozawa, Y.; Mizukami, K. Unique Roles of Rare Variants in the Genetics of Complex Diseases in Humans. J. Hum. Genet. 2021, 66, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Mitani, T.; Isikay, S.; Gezdirici, A.; Gulec, E.Y.; Punetha, J.; Fatih, J.M.; Herman, I.; Akay, G.; Du, H.; Calame, D.G.; et al. High Prevalence of Multilocus Pathogenic Variation in Neurodevelopmental Disorders in the Turkish Population. Am. J. Hum. Genet. 2021, 108, 1981–2005. [Google Scholar] [CrossRef]
- Greally, J.M. Population Epigenetics. Curr. Opin. Syst. Biol. 2017, 1, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the Missing Heritability of Complex Diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef]
- Tyler, A.L.; Crawford, D.C.; Pendergrass, S.A. The Detection and Characterization of Pleiotropy: Discovery, Progress, and Promise. Brief. Bioinform. 2016, 17, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Shikov, A.E.; Skitchenko, R.K.; Predeus, A.V.; Barbitoff, Y.A. Phenome-Wide Functional Dissection of Pleiotropic Effects Highlights Key Molecular Pathways for Human Complex Traits. Sci. Rep. 2020, 10, 1037. [Google Scholar] [CrossRef] [PubMed]
- Schaid, D.J.; Chen, W.; Larson, N.B. From Genome-Wide Associations to Candidate Causal Variants by Statistical Fine-Mapping. Nat. Rev. Genet. 2018, 19, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Hormozdiari, F.; van de Bunt, M.; Segrè, A.V.; Li, X.; Joo, J.W.J.; Bilow, M.; Sul, J.H.; Sankararaman, S.; Pasaniuc, B.; Eskin, E. Colocalization of GWAS and EQTL Signals Detects Target Genes. Am. J. Hum. Genet. 2016, 99, 1245–1260. [Google Scholar] [CrossRef]
- Gu, S.; Olszewski, R.; Nelson, L.; Gallego-Martinez, A.; Lopez-Escamez, J.A.; Hoa, M. Identification of Potential Meniere’s Disease Targets in the Adult Stria Vascularis. Front. Neurol. 2021, 12, 630561. [Google Scholar] [CrossRef]
- Changalidis, A.; Maksiutenko, E.; Barbitoff, Y.; Tkachenko, A.; Vashukova, E.; Pachuliia, O.; Nasykhova, Y.; Glotov, A. Aggregation of Genome-Wide Association Data from FinnGen and UK Biobank Replicates Multiple Risk Loci for Pregnancy Complications. Genes 2022, 13, 2255. [Google Scholar] [CrossRef]
- Fleming, J.S.; McQuillan, H.J.; Millier, M.J.; Sellar, G.C. Expression of Ovarian Tumour Suppressor OPCML in the Female CD-1 Mouse Reproductive Tract. Reproduction 2009, 137, 721–726. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Trait | rsID | Gene | MAF (UKB/FG) | β (UKB/FG) | p-Value (UKB/FG) | Meta p-Value |
|---|---|---|---|---|---|---|
| Ménière disease 1 | rs3777781 | EYA4 | 0.22/0.30 | −0.15/−0.17 | 1.57 × 10−2/2.89 × 10−8 | 1.46 × 10−9 |
| Pre-eclampsia 2 | rs167479 | RGL3 | 0.47/0.42 | 0.06/0.10 | 1.61 × 10−3/1.63 × 10−8 | 1.28 × 10−10 |
| Sexual dysfunction 3 | rs73585022 | OPCML | 5.7 × 10−3/8.3 × 10−4 | 3.68/2.25 | 9.97 × 10−8/8.23 × 10−5 | 1.29 × 10−10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tkachenko, A.A.; Changalidis, A.I.; Maksiutenko, E.M.; Nasykhova, Y.A.; Barbitoff, Y.A.; Glotov, A.S. Replication of Known and Identification of Novel Associations in Biobank-Scale Datasets: A Survey Using UK Biobank and FinnGen. Genes 2024, 15, 931. https://doi.org/10.3390/genes15070931
Tkachenko AA, Changalidis AI, Maksiutenko EM, Nasykhova YA, Barbitoff YA, Glotov AS. Replication of Known and Identification of Novel Associations in Biobank-Scale Datasets: A Survey Using UK Biobank and FinnGen. Genes. 2024; 15(7):931. https://doi.org/10.3390/genes15070931
Chicago/Turabian StyleTkachenko, Alexander A., Anton I. Changalidis, Evgeniia M. Maksiutenko, Yulia A. Nasykhova, Yury A. Barbitoff, and Andrey S. Glotov. 2024. "Replication of Known and Identification of Novel Associations in Biobank-Scale Datasets: A Survey Using UK Biobank and FinnGen" Genes 15, no. 7: 931. https://doi.org/10.3390/genes15070931