
Rare Variants of the SMN1 Gene Detected during Neonatal Screening

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Discrepant Results of Analysis
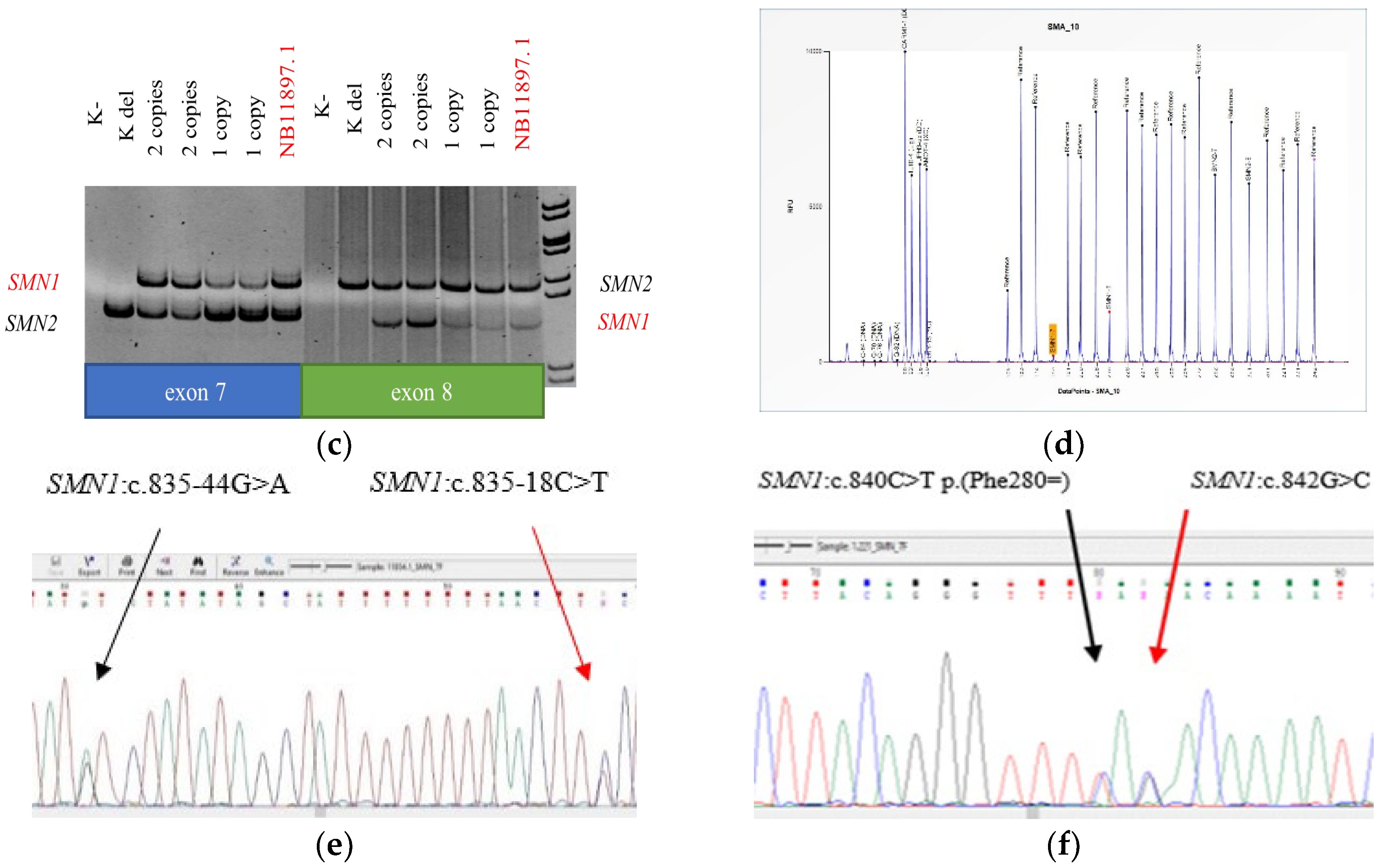
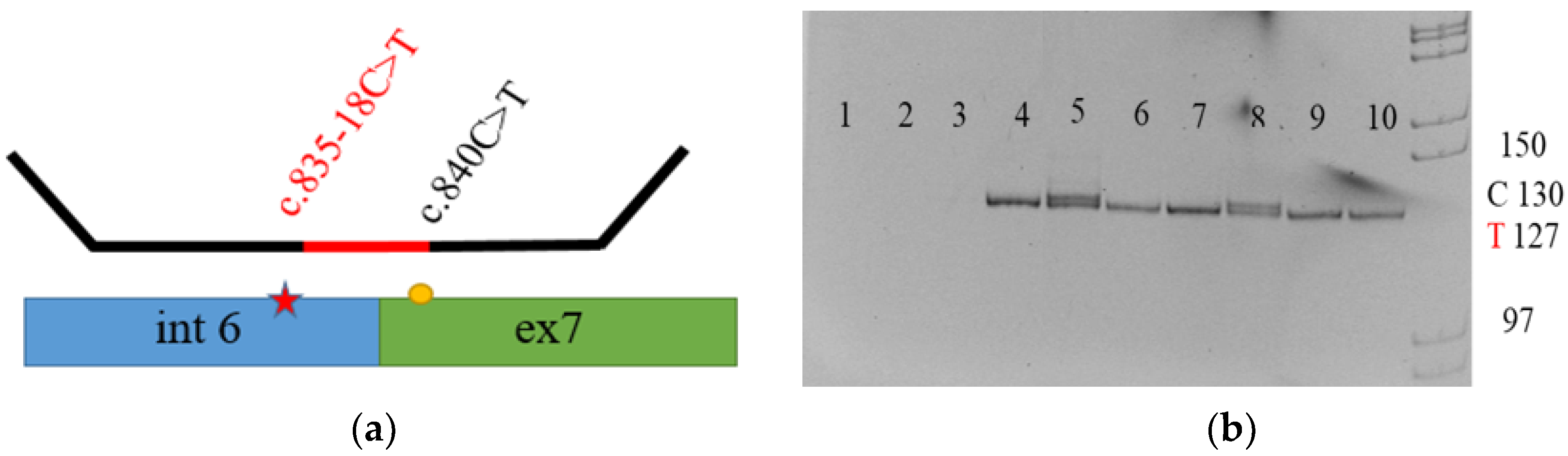
3.1.1. New Variations
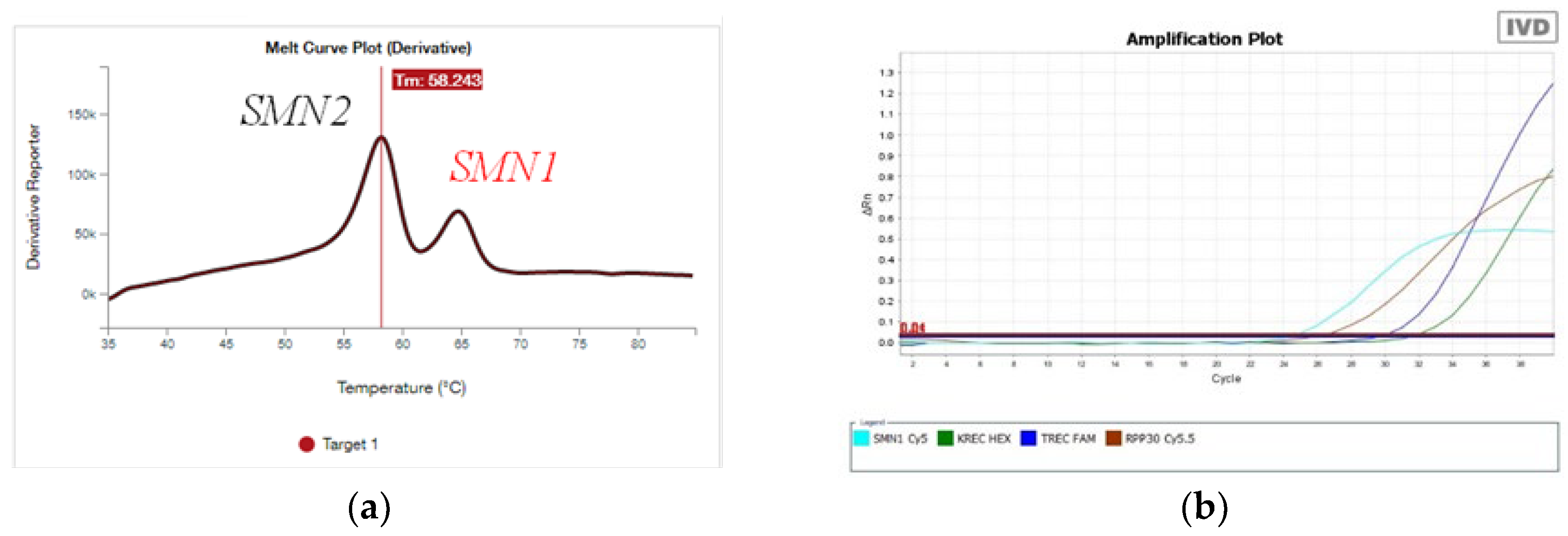
3.1.2. Novel Detection System
3.2. Segregation Analysis
3.3. cDNA Amplification Results
3.4. Analysis of Ethnic Backgrounds of Newborns with Novel Variants
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tisdale, S.; Pellizzoni, L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 2015, 35, 8691–8700. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Butchbach, M.E.R. Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development. Int. J. Mol. Sci. 2021, 22, 7896. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Wilson, R.B. Spinal muscular atrophy: Molecular genetics and diagnostics. Expert Rev. Mol. Diagn. 2004, 4, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Chaytow, H.; Huang, Y.T.; Gillingwater, T.H.; Faller, K.M.E. The role of survival motor neuron protein (SMN) in protein homeostasis. Cell. Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef] [PubMed]
- Voronin, S.; Zakharova, E.; Baydakova, G.; Marakhonov, A.; Shchagina, O.; Ryzhkova, O.; Shilova, N.; Rumyantsev, A.; Shcherbina, A.; Mukhina, A.; et al. Advanced neonatal screening for hereditary diseases in Russia: First results and future prospects. Pediatr. Na GN Speransky 2024, 103, 16–29. [Google Scholar] [CrossRef]
- Zabnenkova, V.V.; Dadali, E.L.; Polyakov, A.V. Proximal spinal muscular atrophy types I–IV: Specific features of molecular genetic diagnosis. Neuromuscul. Dis. 2013, 3, 27–31. (In Russian) [Google Scholar] [CrossRef]
- Zabnenkova, V.V.; Dadali, E.L.; Artemieva, S.B.; Sharkova, I.V.; Rudenskaya, G.E.; Polyakov, A.V. SMN1 gene point mutations in type I–IV proximal spinal muscular atrophy patients with a single copy of SMN1. Russ. J. Genet. 2015, 51, 925–931. [Google Scholar] [CrossRef]
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/495823/ (accessed on 21 April 2024).
- Available online: https://gnomad.broadinstitute.org/gene/ENSG00000172062?dataset=gnomad_r4 (accessed on 22 April 2024).
- Available online: http://ruseq.ru (accessed on 22 April 2024).
- Available online: https://ngs-data-ccu.epigenetic.ru/login/?next=/vcfdb (accessed on 24 April 2024).
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/632986/ (accessed on 25 April 2024).
- Available online: https://gnomad.broadinstitute.org/variant/5-70951948-G-T?dataset=gnomad_r4 (accessed on 27 April 2024).
- Kraszewski, J.N.; Kay, D.M.; Stevens, C.F.; Koval, C.; Haser, B.; Ortiz, V.; Albertorio, A.; Cohen, L.L.; Jain, R.; Andrew, S.P.; et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet. Med. 2018, 20, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Gupta, K.; Ninan, N.S.; Perry, K.; Van Duyne, G.D. The survival motor neuron protein forms soluble glycine zipper oligomers. Structure 2012, 20, 1929–1939. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bowen, B.M.; Truty, R.; Aradhya, S.; Bristow, S.L.; Johnson, B.A.; Morales, A.; Tan, C.A.; Westbrook, M.J.; Winder, T.L.; Chavez, J.C. SMA Identified: Clinical and Molecular Findings from a Sponsored Testing Program for Spinal Muscular Atrophy in More Than 2000 Individuals. Front. Neurol. 2021, 12, 663911. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/633421/ (accessed on 28 April 2024).
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/984453/ (accessed on 28 April 2024).
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/495825/ (accessed on 28 April 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tested | SMN1 (ex7, ex8) | SMN2 (ex7, ex8) | Sequencing (NM_000344.4) |
|---|---|---|---|
| Proband NB11897.1 | 1 copy | 2 copies | c.[835-18C>T];[0] |
| Father NB11897.2 | 2 copies | 2 copies | c.[835-18C>T];[=] |
| Mother NM11897.3 | 1 copy | 2 copies | Not detected |
| Brother NB11897.4 | 2 copies | 2 copies | Not detected |
| Sister NB11897.5 | 1 copy | 2 copies | Not detected |
| Proband NB11907.1 | 1 copy | 3 copies | c.[835-18C>T];[0] |
| Father NB11907.2 | 1 copy | 3 copies | Not detected |
| Mother NB11907.3 | 2 copies | 2 copies | c.[835-18C>T];[=] |
| Proband NB11920.1 | 1 copy | 2 copies | c.[835-18C>T];[0] |
| Father NB11920.2 | 1 copy | 2 copies | Not detected |
| Mother NB11920.3 | 1 copy | 2 copies | c.[835-18C>T];[0] |
| Proband NB11940.1 | 2 copies | 2 copies | c.[835-18C>T];[842G>C] |
| Father NB11940.2 | 1 copy | 3 copies | c.[842G>C];[0] |
| Mother NB11940.3 | 2 copies | 2 copies | c.[835-18C>T];[=] |
| Brother NB11940.4 | 1 copy | 3 copies | Not detected |
| Sister NB11940.5 | 2 copies | 2 copies | c.[835-18C>T];[842G>C] |
| Proband NB1.221 | 1 copy | 2 copies | c.[842G>C];[=] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhkiamova, M.; Polyakov, A.; Marakhonov, A.; Voronin, S.; Saifullina, E.; Vafina, Z.; Michalchuk, K.; Braslavskaya, S.; Chukhrova, A.; Ryadninskaya, N.; et al. Rare Variants of the SMN1 Gene Detected during Neonatal Screening. Genes 2024, 15, 956. https://doi.org/10.3390/genes15070956
Akhkiamova M, Polyakov A, Marakhonov A, Voronin S, Saifullina E, Vafina Z, Michalchuk K, Braslavskaya S, Chukhrova A, Ryadninskaya N, et al. Rare Variants of the SMN1 Gene Detected during Neonatal Screening. Genes. 2024; 15(7):956. https://doi.org/10.3390/genes15070956
Chicago/Turabian StyleAkhkiamova, Maria, Aleksander Polyakov, Andrey Marakhonov, Sergey Voronin, Elena Saifullina, Zulfiia Vafina, Kristina Michalchuk, Svetlana Braslavskaya, Alena Chukhrova, Nina Ryadninskaya, and et al. 2024. "Rare Variants of the SMN1 Gene Detected during Neonatal Screening" Genes 15, no. 7: 956. https://doi.org/10.3390/genes15070956
APA StyleAkhkiamova, M., Polyakov, A., Marakhonov, A., Voronin, S., Saifullina, E., Vafina, Z., Michalchuk, K., Braslavskaya, S., Chukhrova, A., Ryadninskaya, N., Kutsev, S., & Shchagina, O. (2024). Rare Variants of the SMN1 Gene Detected during Neonatal Screening. Genes, 15(7), 956. https://doi.org/10.3390/genes15070956