
Interspecific and Intraspecific Transcriptomic Variations Unveil the Potential High-Altitude Adaptation Mechanisms of the Parnassius Butterfly Species


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
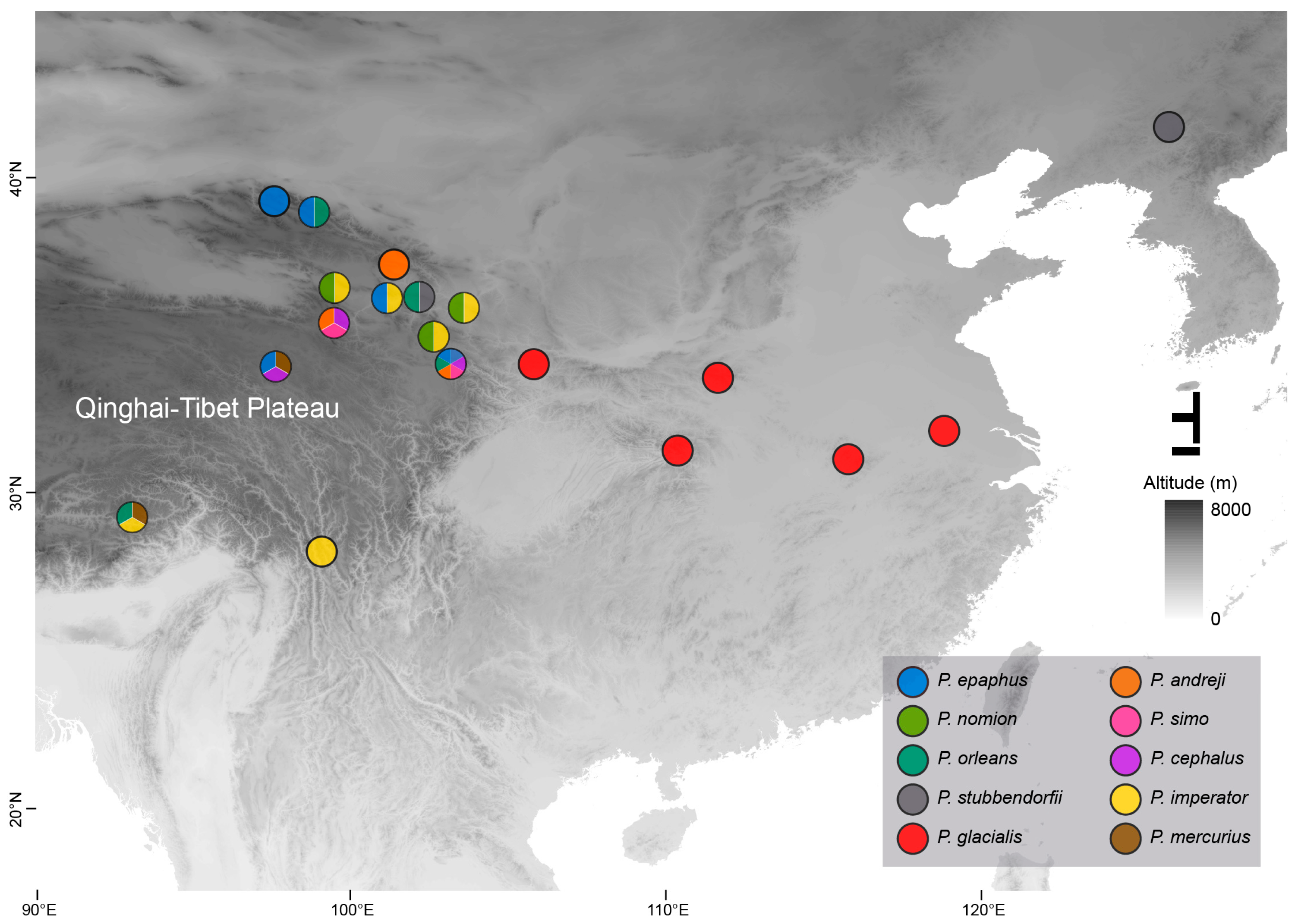
2.1. Sample Collection
2.2. mRNA-Seq Library Construction, Illumina Sequencing, and Gene Abundance Estimation
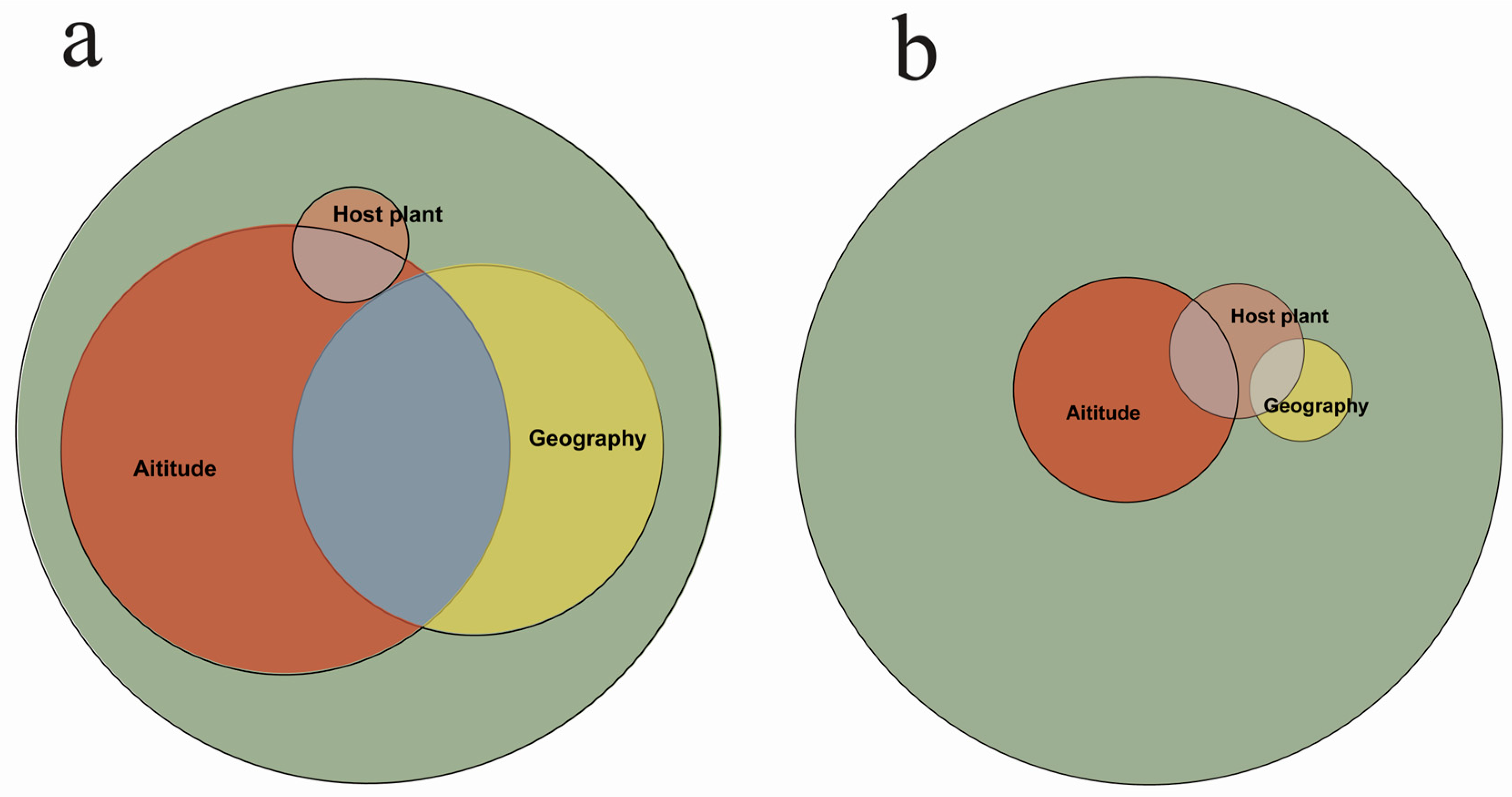
2.3. Multiple Regression Analysis Based on Distance Matrix
2.4. Differential Gene Expression Analysis between Parnassius Species or Intraspecific Populations
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.6. Validation of Gene Expression by RT-qPCR
3. Results
3.1. Transcriptome Sequencing Data Quality
3.2. Multivariate Regression Analysis of Distance Matrices
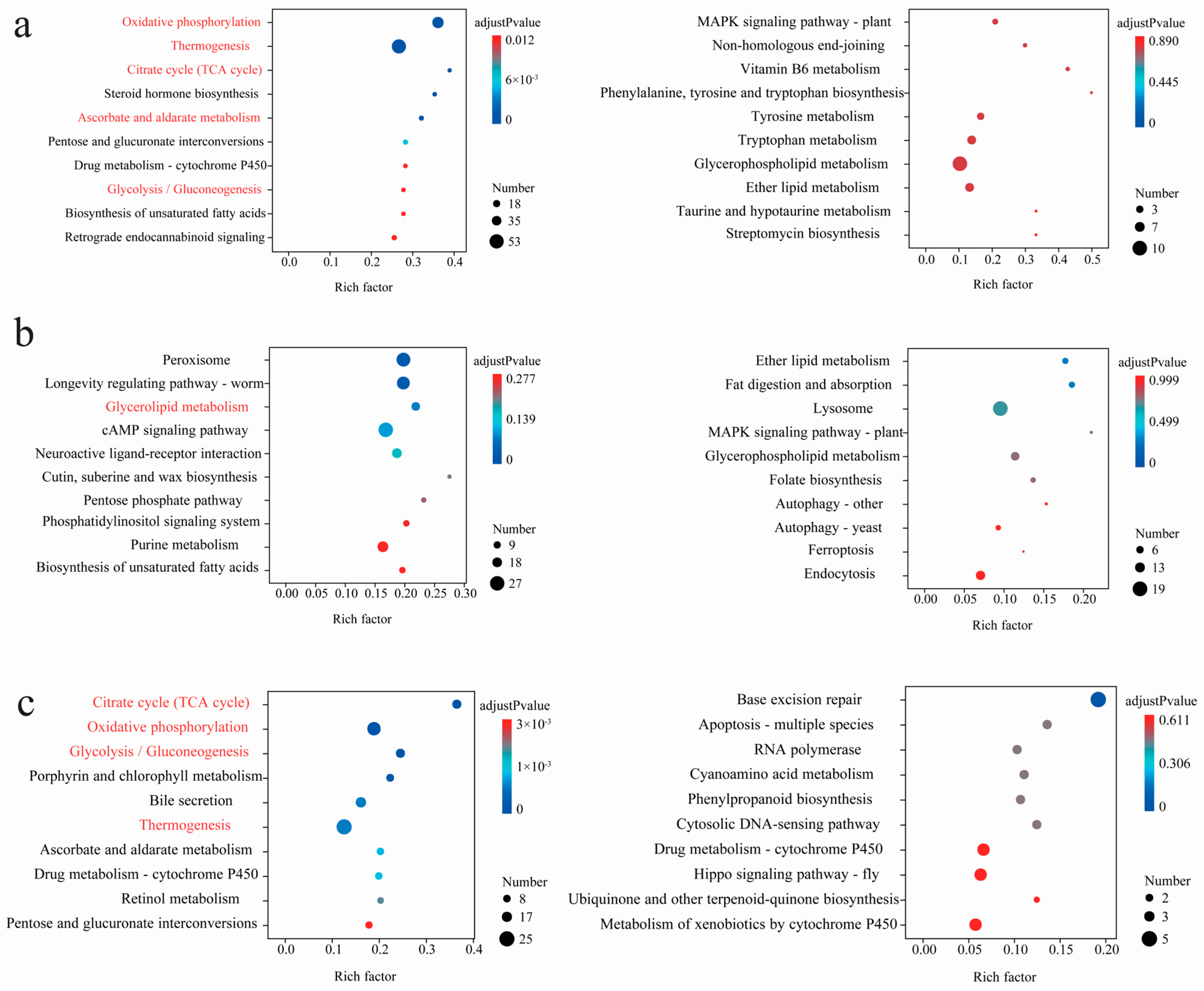
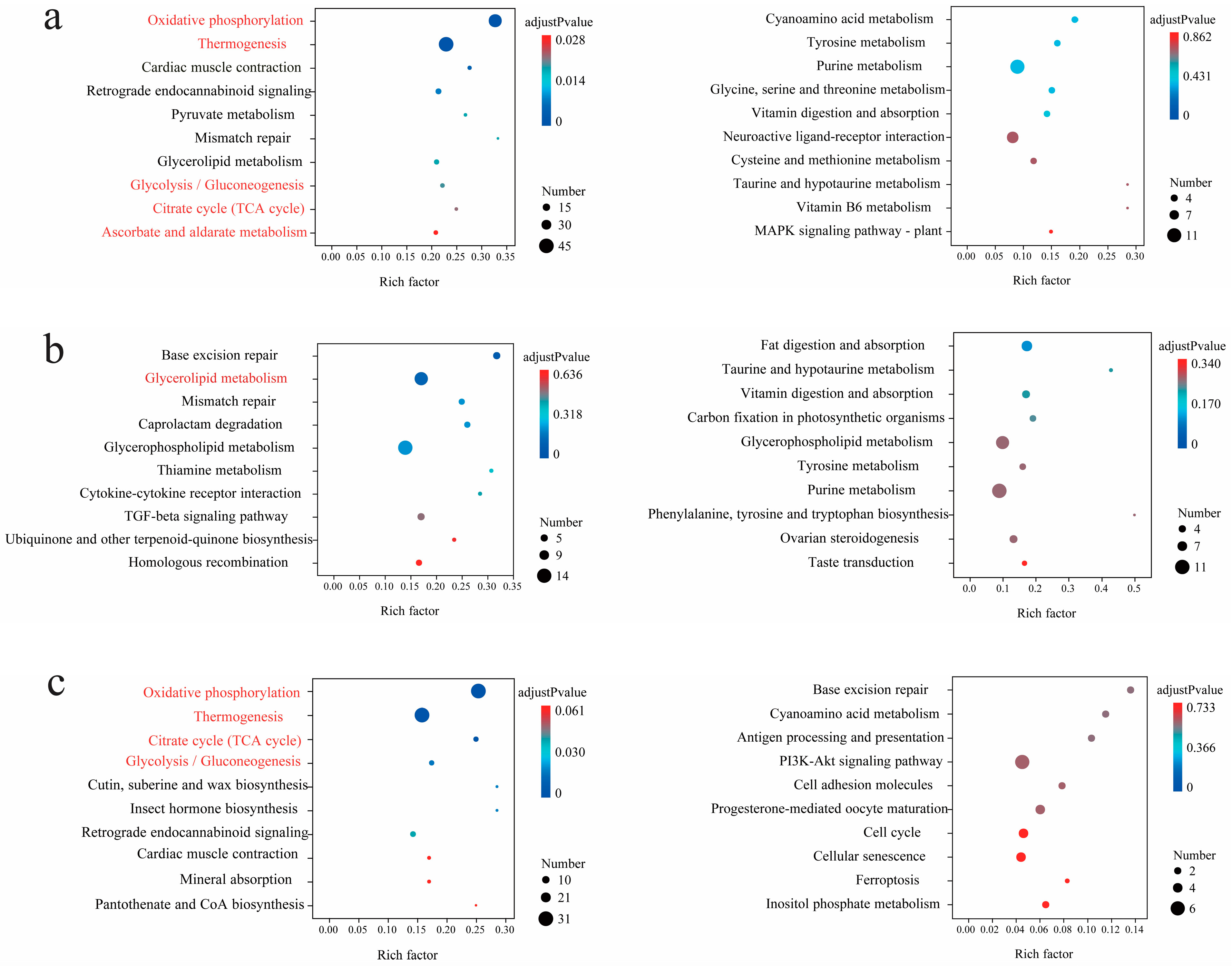
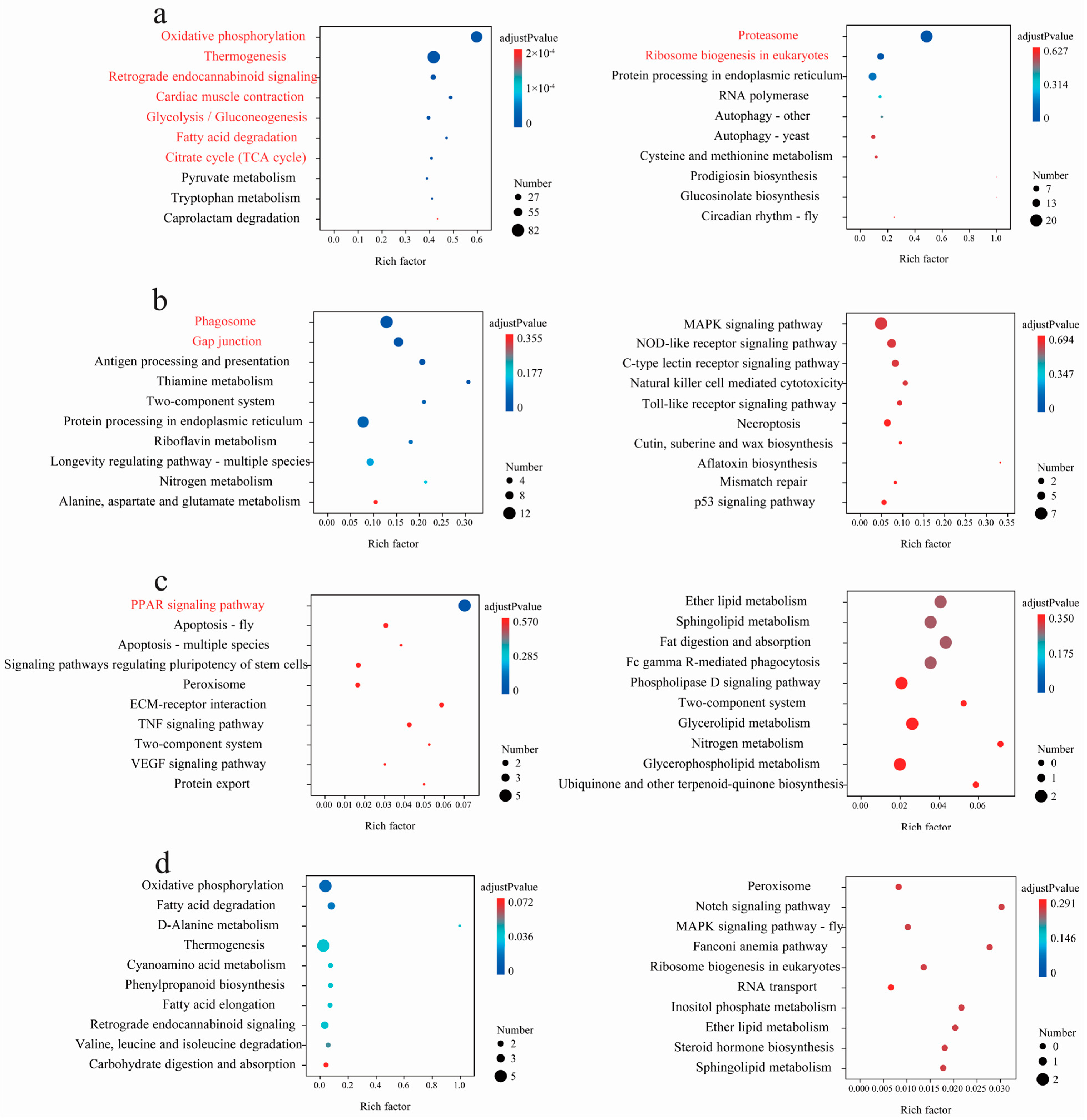
3.3. Statistics of Interspecific Differentially Expressed Genes (DEGs) and Enriched Pathways
3.4. Statistics of Intraspecific Differentially Expressed Genes (DEGs) and Enriched Pathways
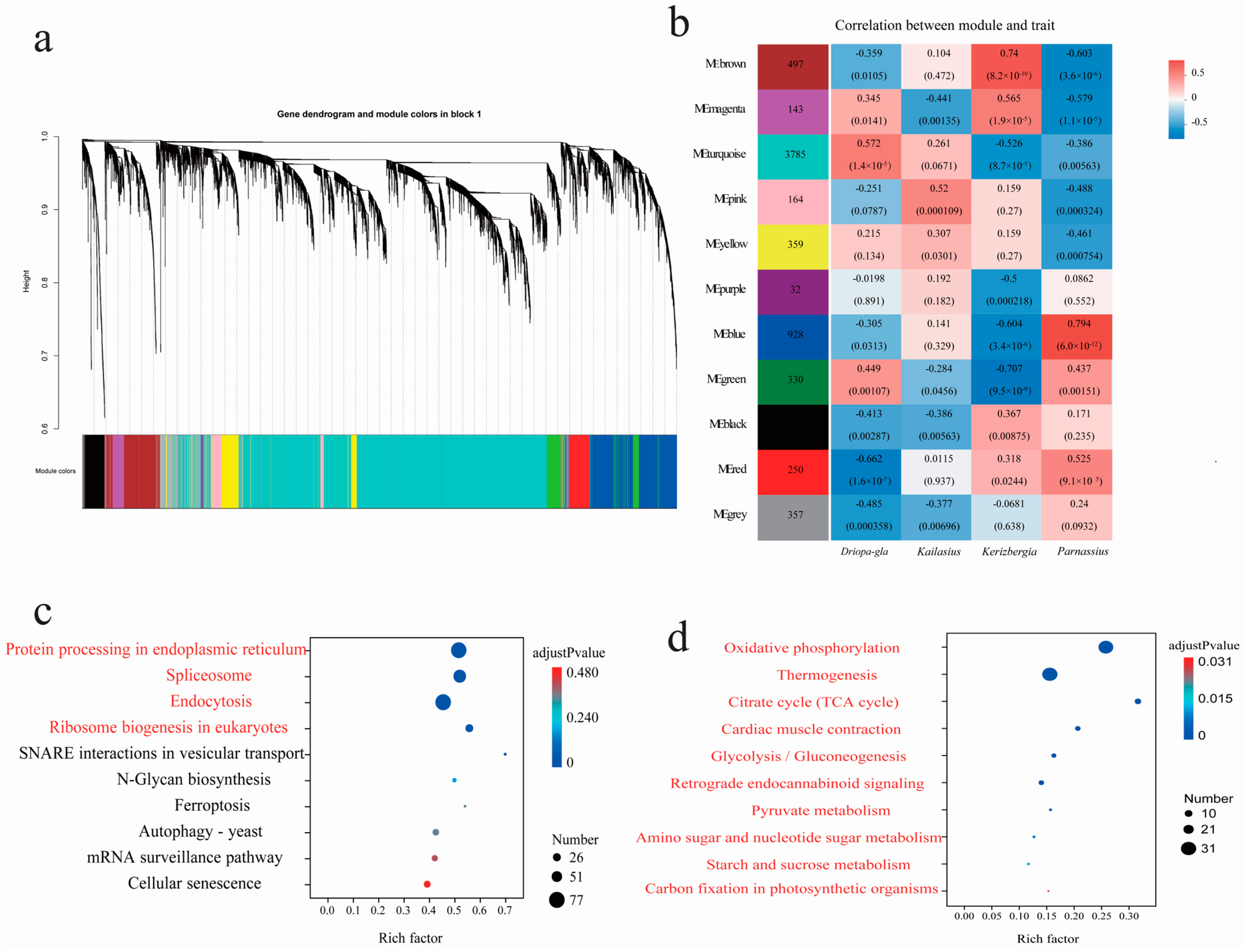
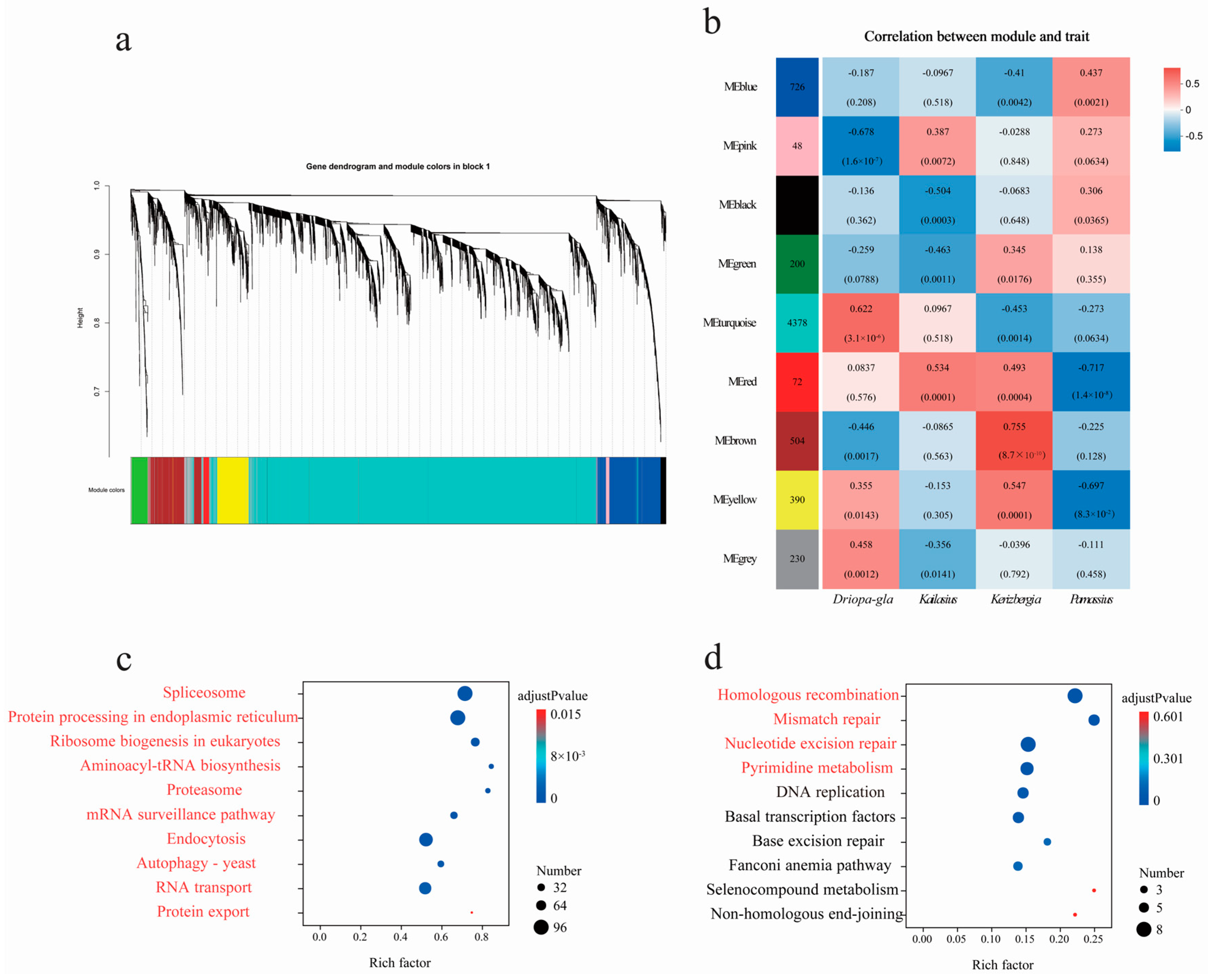
3.5. Featured Modules Based on WGCNA
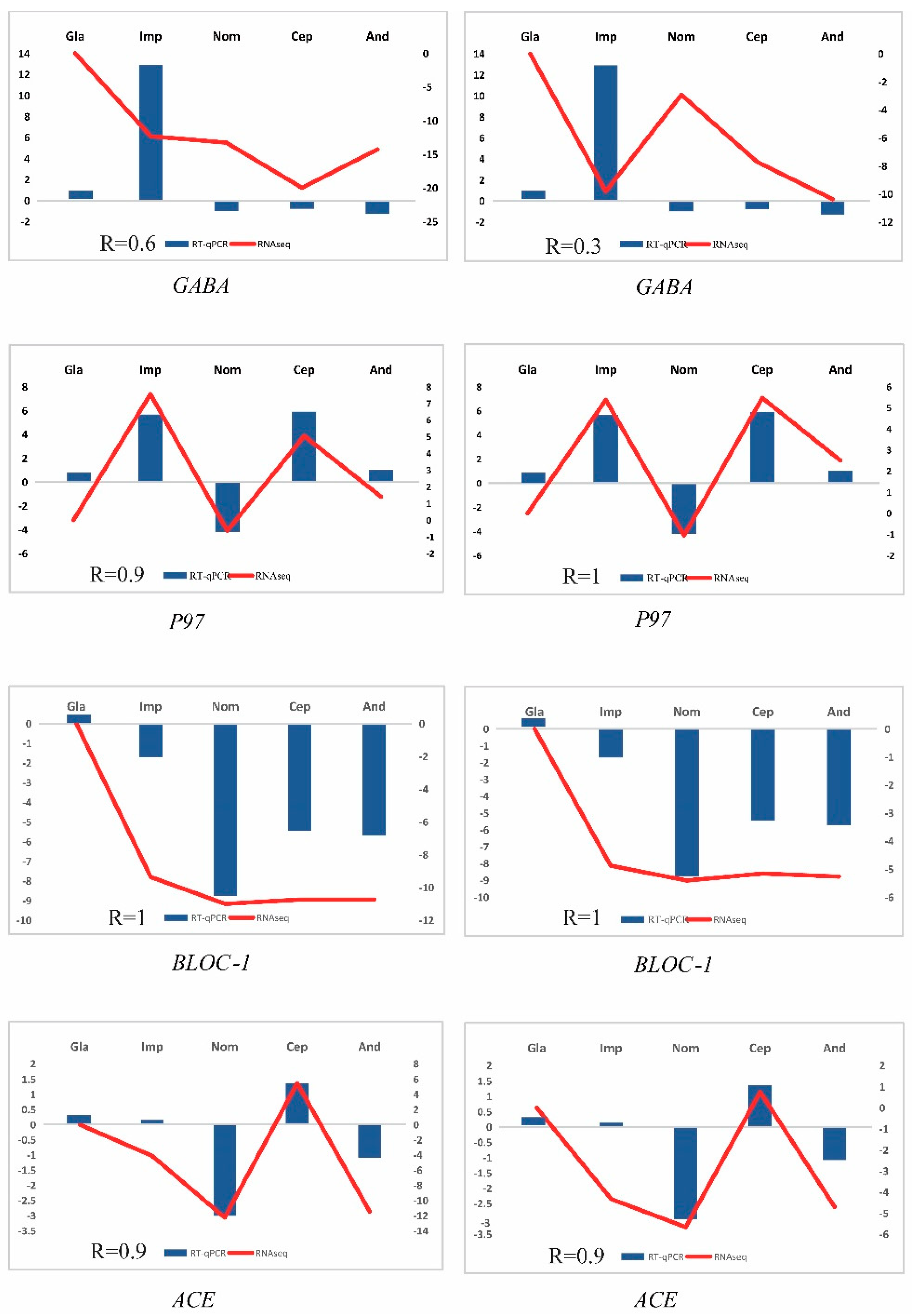
3.6. RNA-Seq Validation Using RT-qPCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Su, C.; Ding, C.; Zhao, Y.; He, B.; Nie, R.; Hao, J. Diapause-linked gene expression pattern and related candidate duplicated genes of the mountain butterfly Parnassius glacialis (Lepidoptera: Papilionidae) revealed by comprehensive transcriptome profiling. Int. J. Mol. Sci. 2023, 24, 5577. [Google Scholar] [CrossRef]
- Ding, D.; Liu, G.; Hou, L.; Gui, W.; Chen, B.; Kang, L. Genetic variation in PTPN1 contributes to metabolic adaptation to high-altitude hypoxia in Tibetan migratory locusts. Nat. Commun. 2018, 9, 4991. [Google Scholar] [CrossRef]
- Zhao, J.; Xia, Y.; Cannon, C.H. Evolutionary diversification of alpine ginger reflects the early uplift of the Himalayan-Tibetan Plateau and rapid extrusion of Indochina. Gondwana Res. 2016, 32, 232–241. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, D.; Zhao, Z.; Zhang, J.; Li, S.; Liu, Z. Transcriptome analysis and identification of genes associated with leaf crude protein content in foxtail millet [Setaria italica (L.) P. Beauv.]. Front. Genet. 2023, 14, 1122212. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, X.; Meng, R.; Liu, C.; Li, M.; Zhang, T. Identification of chemosensory genes from the antennal transcriptome of Semiothisa cinerearia. PLoS ONE 2020, 15, e0237134. [Google Scholar] [CrossRef]
- Zhao, D.J.; Zhang, Z.Y.; Harrison, J. Genome-wide analysis of transcriptional changes in the thoracic muscle of the migratory locust, Locusta migratoria, exposed to hypobaric hypoxia. J. Insect Physiol. 2012, 58, 1424–1431. [Google Scholar] [CrossRef]
- Zhao, D.J.; Zhang, Z.Y.; Cease, A.; Harrison, J.; Kang, J. Efficient utilization of aerobic metabolism helps Tibetan locusts conquer hypoxia. BioMed Cent. Genom. 2013, 14, 631. [Google Scholar] [CrossRef]
- Wu, W.J.; Sun, H.X.; Guo, J.X.; Jiang, F.E.; Liu, X.; Zhang, G.R. De novo transcriptome characterization of the ghost moth, Thitarodes pui, and elevation-based differences in the gene expression of its larvae. Gene 2015, 574, 95–105. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Zhang, L.; Zhao, T.X.; Wang, J.; Zhu, Q.H.; Chen, J.Y.; Yuan, M.L. Gene sequence variations and expression patterns of mitochondrial genes are associated with the adaptive evolution of two Gynaephora species (Lepidoptera: Lymantriinae) living in different high-elevation environments. Gene 2017, 610, 148–155. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, S. Extinction vs. rapid radiation: The juxtaposed evolutionary histories of coelotine spiders support the Eocene–Oligocene orogenesis of the Tibetan Plateau. Syst. Biol. 2017, 66, 988–1006. [Google Scholar] [CrossRef]
- Condamine, F.L. Limited by the roof of the world: Mountain radiations of Apollo swallowtails controlled by diversity-dependence processes. Biol. Lett. 2018, 14, 20170622. [Google Scholar] [CrossRef]
- Keyghobadi, N.; Roland, J.; Strobeck, C. Genetic differentiation and gene flow among populations of the alpine butterfly, Parnassius smintheus, vary with landscape connectivity. Mol. Ecol. 2005, 14, 1897–1909. [Google Scholar] [CrossRef]
- Nazari, V.; Zakharov, E.V.; Sperling, F.A. Phylogeny, historical biogeography, and taxonomic ranking of Parnassiinae (Lepidoptera, Papilionidae) based on morphology and seven genes. Mol. Phylogenetics Evol. 2007, 42, 131–156. [Google Scholar] [CrossRef]
- Schoville, S.D.; Roderick, G.K. Alpine biogeography of Parnassian butterflies during Quaternary climate cycles in North America. Mol. Ecol. 2009, 18, 3471–3485. [Google Scholar] [CrossRef]
- Condamine, F.L.; Sperling, F.A.H.; Kergoat, G.J. Global biogeographical pattern of swallowtail diversification demonstrates alternative colonization routes in the Northern and Southern hemispheres. J. Biogeogr. 2013, 40, 9–23. [Google Scholar] [CrossRef]
- Si, C.; Chen, K.; Tao, R.; Su, C.; Ma, J.; Li, L.; Hao, J.; Yang, Q. Genetic differentiation and divergence time of Chinese Parnassius (Lepidoptera: Papilionidae) species based on nuclear internal transcribed spacer (ITS) sequence data. J. Entomol. Sci. 2020, 55, 520–546. [Google Scholar] [CrossRef]
- Chou, I. Monographia Rhopalocerorum Sinensium; Henan Scientific and Technological Publishing House: Zhengzhou, China, 1994; pp. 191–209. [Google Scholar]
- Tao, R.; Xu, C.; Wang, Y.; Sun, X.; Li, C.; Ma, J.; Hao, J.; Yang, Q. Spatiotemporal differentiation of alpine butterfly Parnassius glacialis (Papilionidae: Parnassiinae) in China: Evidence from mitochondrial DNA and nuclear single nucleotide polymorphisms. Genes 2020, 11, 188. [Google Scholar] [CrossRef]
- Allio, R.; Nabholz, B.; Wanke, S.; Chomicki, G.; Pérez-Escobar, O.A.; Cotton, A.M.; Clamens, A.-L.; Kergoat, G.J.; Sperling, F.A.; Condamine, F.L. Genome-wide macroevolutionary signatures of key innovations in butterflies colonizing new host plants. Nat. Commun. 2021, 12, 354. [Google Scholar] [CrossRef]
- He, B.; Zhao, Y.J.; Su, C.Y.; Lin, G.H.; Wang, Y.L.; Li, L.Y.; Ma, J.Y.; Yang, Q.; Hao, J.S. Phylogenomics reveal extensive phylogenetic discordance due to incomplete lineage sorting following the rapid radiation of alpine butterflies (Papilionidae: Parnassius). Syst. Entomol. 2023, 48, 585–599. [Google Scholar] [CrossRef]
- Su, C.Y.; Xie, T.T.; Wang, Y.L.; Si, C.C.; Li, L.Y.; Ma, J.Y.; Li, C.X.; Sun, X.Y.; Hao, J.S.; Yang, Q. Miocene diversification and high-altitude adaptation of Parnassius butterflies (Lepidoptera: Papilionidae) in Qinghai–Tibet plateau revealed by large-scale transcriptomic data. Insects 2020, 11, 754. [Google Scholar] [CrossRef]
- Zhao, Y.J.; He, B.; Tao, R.S.; Su, C.Y.; Ma, J.Y.; Hao, J.S.; Yang, Q. Phylogeny and biogeographic history of Parnassius butterflies (Papilionidae: Parnassiinae) reveal their origin and deep diversification in West China. Insects 2022, 13, 406. [Google Scholar] [CrossRef]
- Zhao, Y.; Su, C.; He, B.; Nie, R.; Wang, Y.; Ma, J.; Song, J.; Yang, Q.; Hao, J. Dispersal from the Qinghai-Tibet plateau by a high-altitude butterfly is associated with rapid expansion and reorganization of its genome. Nat. Commun. 2023, 14, 8190. [Google Scholar] [CrossRef]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Lichstein, J.W. Multiple regression on distance matrices: A multivariate spatial analysis tool. J. Plant Ecol. 2007, 188, 117–131. [Google Scholar] [CrossRef]
- Goslee, S.C.; Urban, D.L. The ecodist package for dissimilarity-based analysis of ecological data. J. Stat. Softw. 2007, 22, 1–19. [Google Scholar] [CrossRef]
- Weinstein, S.B.; Martínez-Mota, R.; Stapleton, T.E.; Klure, D.M.; Greenhalgh, R.; Orr, T.J.; Dale, C.; Kohl, K.D.; Dearing, M.D. Microbiome stability and structure is governed by host phylogeny over diet and geography in woodrats (Neotoma spp.). Proc. Natl. Acad. Sci. USA 2021, 118, e2108787118. [Google Scholar] [CrossRef]
- Liu, H.; Wang, C.; Qiu, C.L.; Shi, J.H.; Sun, Z.; Hu, X.J.; Liu, L.; Wang, M.Q. A salivary odorant-binding protein mediates Nilaparvata lugens feeding and host plant phytohormone suppression. Int. J. Mol. Sci. 2021, 22, 4988. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Curr. Bioinform. 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Chu, C.; Wang, S.; Chen, L.; Lu, J.; Kong, X.; Huang, T.; Li, H.; Cai, Y.D. The use of Gene Ontology term and KEGG pathway enrichment for analysis of drug half-life. PLoS ONE 2016, 11, e0165496. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, G.; Shi, C.; Liu, L.; Guo, Q.; Han, C.; Zhang, D.; Zhang, L.; Liu, B.; Gao, H. Majorbio Cloud: A one-stop, comprehensive bioinformatic platform for multiomics analyses. IMeta 2022, 1, e12. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pan, H.; Yang, X.; Bidne, K.; Hellmich, R.L.; Siegfried, B.D.; Zhou, X. Selection of reference genes for RT-qPCR analysis in the monarch butterfly, Danaus plexippus (L.), a migrating bio-indicator. PLoS ONE 2015, 10, e0129482. [Google Scholar] [CrossRef]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef]
- Takahashi, N.; Goto, T.; Kusudo, T.; Moriyama, T.; Kawada, T. The structures and functions of peroxisome proliferator-activated receptors (PPARs). Nihon Rinsho. Jpn. J. Clin. Med. 2005, 63, 557–564. [Google Scholar]
- Fang, L.; Zhang, M.; Li, Y.; Liu, Y.; Cui, Q.; Wang, N. PPARgene: A database of experimentally verified and computationally predicted PPAR target genes. PPAR Res. 2016, 2016, 6042162. [Google Scholar] [CrossRef]
- Cui, M.; Hu, P.; Wang, T.; Tao, J.; Zong, S. Differential transcriptome analysis reveals genes related to cold tolerance in seabuckthorn carpenter moth, Eogystia hippophaecolus. PLoS ONE 2017, 12, e0187105. [Google Scholar] [CrossRef]
- Galen, S.C.; Natarajan, C.; Moriyama, H.; Weber, R.E.; Fago, A.; Benham, P.M.; Chavez, A.N.; Cheviron, Z.A.; Storz, J.F.; Witt, C.C. Contribution of a mutational hot spot to hemoglobin adaptation in high-altitude Andean house wrens. Proc. Natl. Acad. Sci. USA 2015, 112, 13958–13963. [Google Scholar] [CrossRef]
- Natarajan, C.; Hoffmann, F.G.; Weber, R.E.; Fago, A.; Witt, C.C.; Storz, J.F. Predictable convergence in hemoglobin function has unpredictable molecular underpinnings. Science 2016, 354, 336–339. [Google Scholar] [CrossRef]
- Nath, S.; Villadsen, J. Oxidative phosphorylation revisited. Biotechnol. Bioeng. 2015, 112, 429–437. [Google Scholar] [CrossRef]
- Wierenga, R.; Kapetaniou, E.; Venkatesan, R. Triosephosphate isomerase: A highly evolved biocatalyst. Cell. Mol. Life Sci. 2010, 67, 3961–3982. [Google Scholar] [CrossRef]
- Li, Q.; Liu, X.; Li, L.; Ge, C.; Jian, L. A comprehensive analysis of hepatopancreas metabolomics and transcriptomics provides insights into the growth of three-year-old crabs (Eriocheir sinensis) under low temperature. Comp. Biochem. Physiol. Part D Genom. Proteom. 2024, 49, 101182. [Google Scholar] [CrossRef]
- Kressler, D.; Hurt, E.; Baβler, J. Driving ribosome assembly. BBA-Mol. Cell Res. 2010, 1803, 673–683. [Google Scholar] [CrossRef]
- Storz, J.F.; Scott, G.R. Life ascending: Mechanism and process in physiological adaptation to high-altitude hypoxia. Annu. Rev. Ecol. Evol. Syst. 2019, 50, 503–526. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, C.; Su, C.; Li, Y.; Zhao, Y.; Wang, Y.; Wang, Y.; Nie, R.; He, B.; Ma, J.; Hao, J. Interspecific and Intraspecific Transcriptomic Variations Unveil the Potential High-Altitude Adaptation Mechanisms of the Parnassius Butterfly Species. Genes 2024, 15, 1013. https://doi.org/10.3390/genes15081013
Ding C, Su C, Li Y, Zhao Y, Wang Y, Wang Y, Nie R, He B, Ma J, Hao J. Interspecific and Intraspecific Transcriptomic Variations Unveil the Potential High-Altitude Adaptation Mechanisms of the Parnassius Butterfly Species. Genes. 2024; 15(8):1013. https://doi.org/10.3390/genes15081013
Chicago/Turabian StyleDing, Chen, Chengyong Su, Yali Li, Youjie Zhao, Yunliang Wang, Ying Wang, Ruie Nie, Bo He, Junye Ma, and Jiasheng Hao. 2024. "Interspecific and Intraspecific Transcriptomic Variations Unveil the Potential High-Altitude Adaptation Mechanisms of the Parnassius Butterfly Species" Genes 15, no. 8: 1013. https://doi.org/10.3390/genes15081013
APA StyleDing, C., Su, C., Li, Y., Zhao, Y., Wang, Y., Wang, Y., Nie, R., He, B., Ma, J., & Hao, J. (2024). Interspecific and Intraspecific Transcriptomic Variations Unveil the Potential High-Altitude Adaptation Mechanisms of the Parnassius Butterfly Species. Genes, 15(8), 1013. https://doi.org/10.3390/genes15081013