
Cardiac Phenotype and Gene Mutations in RASopathies

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Molecular Involvement of RAS/MAPK Signaling Pathway in the Development of Heart Diseases
3. Heart Diseases Associated with RASopathies
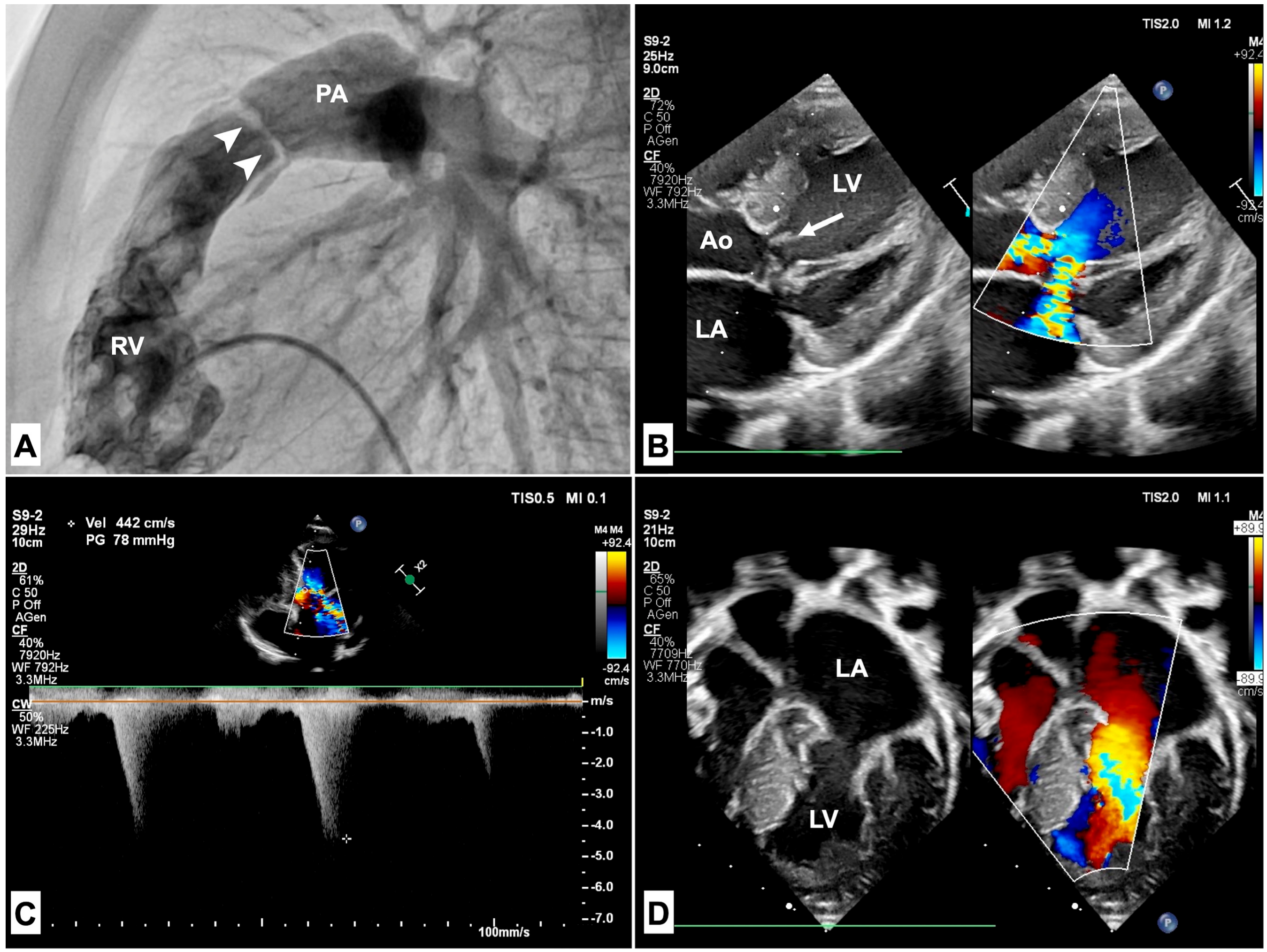
3.1. Pulmonary Stenosis
3.2. Hypertrophic Cardiomyopathy
4. Discussion
5. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Tartaglia, M.; Aoki, Y.; Gelb, B.D. The molecular genetics of RASopathies: An update on novel disease genes and new disorders. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Zenker, M. Clinical overview on RASopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Noonan, J.A. Hypertelorism with Turner Phenotype: A New Syndrome with Associated Congenital Heart Disease. Am. J. Dis. Child. 1968, 116, 373–380. [Google Scholar] [CrossRef]
- Roberts, A.E. Noonan Syndrome. In Gene Reviews((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 1993. [Google Scholar]
- Libraro, A.; D’Ascanio, V.; Cappa, M.; Chiarito, M.; Digilio, M.C.; Einaudi, S.; Grandone, A.; Maghnie, M.; Mazzanti, L.; Mussa, A.; et al. Growth in Children with Noonan Syndrome and Effects of Growth Hormone Treatment on Adult Height. Front. Endocrinol. 2021, 12, 761171. [Google Scholar] [CrossRef]
- Johnston, J.J.; van der Smagt, J.J.; Rosenfeld, J.A.; Pagnamenta, A.T.; Alswaid, A.; Baker, E.H.; Blair, E.; Borck, G.; Brinkmann, J.; Craigen, W.; et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet. Med. 2018, 20, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- van der Burgt, I. Noonan syndrome. Orphanet J. Rare Dis. 2007, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Leoni, C.; Blandino, R.; Delogu, A.B.; De Rosa, G.; Onesimo, R.; Verusio, V.; Marino, M.V.; Lanza, G.A.; Rigante, D.; Tartaglia, M.; et al. Genotype-cardiac phenotype correlations in a large single-center cohort of patients affected by RASopathies: Clinical implications and literature review. Am. J. Med. Genet. Part A 2021, 188, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Asmamaw, M.D.; Shi, X.-J.; Zhang, L.-R.; Liu, H.-M. A comprehensive review of SHP2 and its role in cancer. Cell. Oncol. 2022, 45, 729–753. [Google Scholar] [CrossRef]
- Lepri, F.; De Luca, A.; Stella, L.; Rossi, C.; Baldassarre, G.; Pantaleoni, F.; Cordeddu, V.; Williams, B.J.; Dentici, M.L.; Caputo, V.; et al. SOS1 mutations in Noonan syndrome: Molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations. Hum. Mutat. 2011, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Faienza, M.F.; Giordani, L.; Ferraris, M.; Bona, G.; Cavallo, L. PTPN11 gene mutation and severe neonatal hypertrophic cardiomyopathy: What is the link? Pediatr. Cardiol. 2009, 30, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Calcagni, G.; Limongelli, G.; D’Ambrosio, A.; Gesualdo, F.; Digilio, M.C.; Baban, A.; Albanese, S.B.; Versacci, P.; De Luca, E.; Ferrero, G.B.; et al. Cardiac defects, morbidity and mortality in patients affected by RASopathies. CARNET study results. Int. J. Cardiol. 2017, 245, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Hilal, N.; Chen, Z.; Chen, M.H.; Choudhury, S. RASopathies and cardiac manifestations. Front. Cardiovasc. Med. 2023, 10, 1176828. [Google Scholar] [CrossRef] [PubMed]
- Calcagni, G.; Gagliostro, G.; Limongelli, G.; Unolt, M.; De Luca, E.; Digilio, M.C.; Baban, A.; Albanese, S.B.; Ferrero, G.B.; Baldassarre, G.; et al. Atypical cardiac defects in patients with RASopathies: Updated data on CARNET study. Birth Defects Res. 2020, 112, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Gelb, B.D.; Roberts, A.E.; Tartaglia, M. Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog. Pediatr. Cardiol. 2015, 39, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Lauriol, J.; Jaffré, F.; Kontaridis, M.I. The role of the protein tyrosine phosphatase SHP2 in cardiac development and disease. Semin. Cell Dev. Biol. 2015, 37, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.; Suñé, G.; Faucherre, A.; Fabregat, C.; Izpisua Belmonte, J.C. Hypoxia Induces Myocardial Regeneration in Zebrafish. Circulation 2012, 126, 3017–3027. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Chan, G.; Newbigging, S.; Morikawa, L.; Bronson, R.T.; Neel, B.G. Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc. Natl. Acad. Sci. USA 2009, 106, 4736–4741. [Google Scholar] [CrossRef] [PubMed]
- Schramm, C.; Fine, D.M.; Edwards, M.A.; Reeb, A.N.; Krenz, M. The PTPN11 loss-of-function mutation Q510E-Shp2 causes hypertrophic cardiomyopathy by dysregulating mTOR signaling. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, H231–H243. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Lauriol, J.; Thul, J.; Behnke-Hall, K.; Logeswaran, T.; Schanzer, A.; Bogurcu, N.; Garvalov, B.K.; Zenker, M.; Gelb, B.D.; et al. Rapidly progressive hypertrophic cardiomyopathy in an infant with Noonan syndrome with multiple lentigines: Palliative treatment with a rapamycin analog. Am. J. Med. Genet. Part A 2015, 167, 744–751. [Google Scholar] [CrossRef]
- Ramos-Kuri, M.; Meka, S.H.; Salamanca-Buentello, F.; Hajjar, R.J.; Lipskaia, L.; Chemaly, E.R. Molecules linked to Ras signaling as therapeutic targets in cardiac pathologies. Biol. Res. 2021, 54, 23. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Porras, I.; Fabbiano, S.; Schuhmacher, A.J.; Aicher, A.; Canamero, M.; Camara, J.A.; Cusso, L.; Desco, M.; Heeschen, C.; Mulero, F.; et al. K-RasV14I recapitulates Noonan syndrome in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 16395–16400. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Simpson, J.; Hong, J.H.; Kim, K.H.; Thavarajah, N.K.; Backx, P.H.; Neel, B.G.; Araki, T. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J. Clin. Investig. 2011, 121, 1009–1025. [Google Scholar] [CrossRef] [PubMed]
- Takahara, S.; Inoue, S.I.; Miyagawa-Tomita, S.; Matsuura, K.; Nakashima, Y.; Niihori, T.; Matsubara, Y.; Saiki, Y.; Aoki, Y. New Noonan syndrome model mice with RIT1 mutation exhibit cardiac hypertrophy and susceptibility to β-adrenergic stimulation-induced cardiac fibrosis. eBioMedicine 2019, 42, 43–53. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Ryu, M.J.; Cho, E.; Sang, Y.; Damnernsawad, A.; Zhou, Y.; Liu, Y.; Zhang, J.; Lee, Y. Embryonic Expression of Nras(G 12 D) Leads to Embryonic Lethality and Cardiac Defects. Front. Cell Dev. Biol. 2021, 9, 633661. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Wakimoto, H.; Conner, D.; Araki, T.; Yuan, T.; Roberts, A.; Seidman, C.; Bronson, R.; Neel, B.; Seidman, J.G.; et al. Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome-associated Sos1 mutation. J. Clin. Investig. 2010, 120, 4353–4365. [Google Scholar] [CrossRef]
- Inoue, S.-I.; Moriya, M.; Watanabe, Y.; Miyagawa-Tomita, S.; Niihori, T.; Oba, D.; Ono, M.; Kure, S.; Ogura, T.; Matsubara, Y.; et al. New BRAF knockin mice provide a pathogenetic mechanism of developmental defects and a therapeutic approach in cardio-facio-cutaneous syndrome. Hum. Mol. Genet. 2014, 23, 6553–6566. [Google Scholar] [CrossRef] [PubMed]
- Motta, M.; Giancotti, A.; Mastromoro, G.; Chandramouli, B.; Pinna, V.; Pantaleoni, F.; Di Giosaffatte, N.; Petrini, S.; Mazza, T.; D’Ambrosio, V.; et al. Clinical and functional characterization of a novel RASopathy-causing SHOC2 mutation associated with prenatal-onset hypertrophic cardiomyopathy. Hum. Mutat. 2019, 40, 1046–1056. [Google Scholar] [PubMed]
- Lam, J.; Corno, A.; Oorthuys, H.W.E.; Marcelletti, C. Unusual combination of congenital heart lesions in a child with Noonan’s syndrome. Pediatr. Cardiol. 1982, 3, 23–26. [Google Scholar] [CrossRef]
- Lawrence, K.M.; Burstein, D.S.; Ahrens-Nicklas, R.; Gaynor, J.W.; Nuri, M.A. Noonan syndrome associated with hypoplastic left heart syndrome. Cardiol. Young 2023, 33, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.C.; Conti, E.; Sarkozy, A.; Mingarelli, R.; Dottorini, T.; Marino, B.; Pizzuti, A.; Dallapiccola, B. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am. J. Hum. Genet. 2002, 71, 389–394. [Google Scholar] [CrossRef]
- Alexander, P.M.A.; Nugent, A.W.; Daubeney, P.E.F.; Lee, K.J.; Sleeper, L.A.; Schuster, T.; Turner, C.; Davis, A.M.; Semsarian, C.; Colan, S.D.; et al. Long-Term Outcomes of Hypertrophic Cardiomyopathy Diagnosed during Childhood: Results from a National Population-Based Study. Circulation 2018, 138, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Pacileo, G.; Marino, B.; Digilio, M.C.; Sarkozy, A.; Elliott, P.; Versacci, P.; Calabro, P.; De Zorzi, A.; Di Salvo, G.; et al. Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. Am. J. Cardiol. 2007, 100, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Prendiville, T.W.; Gauvreau, K.; Tworog-Dube, E.; Patkin, L.; Kucherlapati, R.S.; Roberts, A.E.; Lacro, R.V. Cardiovascular disease in Noonan syndrome. Arch. Dis. Child. 2014, 99, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Sreeram, N.; Kitchiner, D.; Smith, A. Spectrum of valvar abnormalities in Noonan’s syndrome—A pathologic study. Cardiol. Young 2008, 4, 62–66. [Google Scholar] [CrossRef]
- Shaw, A.C.; Kalidas, K.; Crosby, A.H.; Jeffery, S.; Patton, M.A. The natural history of Noonan syndrome: A long-term follow-up study. Arch. Dis. Child. 2007, 92, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Delogu, A.B.; Limongelli, G.; Versacci, P.; Adorisio, R.; Kaski, J.P.; Blandino, R.; Maiolo, S.; Monda, E.; Putotto, C.; De Rosa, G.; et al. The heart in RASopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, A.; Conti, E.; Seripa, D.; Digilio, M.C.; Grifone, N.; Tandoi, C.; Fazio, V.M.; Di Ciommo, V.; Marino, B.; Pizzuti, A.; et al. Correlation between PTPN11 gene mutations and congenital heart defects in Noonan and LEOPARD syndromes. J. Med. Genet. 2003, 40, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Pennacchio, L.A.; Zhao, C.; Yadav, K.K.; Fodale, V.; Sarkozy, A.; Pandit, B.; Oishi, K.; Martinelli, S.; Schackwitz, W.; et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet. 2007, 39, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Cordeddu, V.; Yin, J.C.; Gunnarsson, C.; Virtanen, C.; Drunat, S.; Lepri, F.; De Luca, A.; Rossi, C.; Ciolfi, A.; Pugh, T.J.; et al. Activating Mutations Affecting the Dbl Homology Domain of SOS2 Cause Noonan Syndrome. Hum. Mutat. 2015, 36, 1080–1087. [Google Scholar] [CrossRef]
- Lin, A.E.; Basson, C.T.; Goldmuntz, E.; Magoulas, P.L.; McDermott, D.A.; McDonald-McGinn, D.M.; McPherson, E.; Morris, C.A.; Noonan, J.; Nowak, C.; et al. Adults with genetic syndromes and cardiovascular abnormalities: Clinical history and management. Genet. Med. 2008, 10, 469–494. [Google Scholar] [CrossRef] [PubMed]
- McCrindle, B.W. Independent predictors of long-term results after balloon pulmonary valvuloplasty. Valvuloplasty and Angioplasty of Congenital Anomalies (VACA) Registry Investigators. Circulation 1994, 89, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, P.; Dearani, J.A.; Daly, R.C.; King, K.S.; Ammash, N.M.; Cetta, F.; Schaff, H.V. Early Outcomes of Cardiac Surgery in Patients with Noonan Syndrome. Semin. Thorac. Cardiovasc. Surg. 2019, 31, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Masura, J.; Burch, M.; Deanfield, J.E.; Sullivan, I.D. Five-year follow-up after balloon pulmonary valvuloplasty. J. Am. Coll. Cardiol. 1993, 21, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, J.; Tibby, S.M.; Rosenthal, E.; Qureshi, S.; Morgan, G.; Krasemann, T. Results of balloon pulmonary valvoplasty in children with Noonan’s syndrome. Cardiol. Young 2018, 28, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Abumehdi, M.; Mehta, C.; Afifi, A.; Yong, S.F.; Chaudhari, M.; Bhole, V.; Dhillon, R.; Stumper, O. Supravalvular pulmonary stenosis: A risk factor for reintervention in Noonan syndrome with pulmonary valve stenosis. Catheter. Cardiovasc. Interv. 2022, 99, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, M.E.; Digilio, M.C. Cardiovascular disease in Noonan syndrome. Curr. Opin. Pediatr. 2018, 30, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, H.; De Backer, J.; Babu-Narayan, S.V.; Budts, W.; Chessa, M.; Diller, G.P.; Lung, B.; Kluin, J.; Lang, I.M.; Meijboom, F.; et al. 2020 ESC Guidelines for the management of adult congenital heart disease. Eur. Heart J. 2021, 42, 563–645. [Google Scholar] [CrossRef] [PubMed]
- Norrish, G.; Field, E.; McLeod, K.; Ilina, M.; Stuart, G.; Bhole, V.; Uzun, O.; Brown, E.; Daubeney, P.E.F.; Lota, A.; et al. Clinical presentation and survival of childhood hypertrophic cardiomyopathy: A retrospective study in United Kingdom. Eur. Heart J. 2019, 40, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Rubino, M.; Lioncino, M.; Di Fraia, F.; Pacileo, R.; Verrillo, F.; Cirillo, A.; Caiazza, M.; Fusco, A.; Esposito, A.; et al. Hypertrophic Cardiomyopathy in Children: Pathophysiology, Diagnosis, and Treatment of Non-sarcomeric Causes. Front. Pediatr. 2021, 9, 632293. [Google Scholar] [CrossRef] [PubMed]
- Calcagni, G.; Adorisio, R.; Martinelli, S.; Grutter, G.; Baban, A.; Versacci, P.; Digilio, M.C.; Drago, F.; Gelb, B.D.; Tartaglia, M.; et al. Clinical Presentation and Natural History of Hypertrophic Cardiomyopathy in RASopathies. Heart Fail. Clin. 2018, 14, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Gelb, B.D.; Tartaglia, M. RAS signaling pathway mutations and hypertrophic cardiomyopathy: Getting into and out of the thick of it. J. Clin. Investig. 2011, 121, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, F.; Pacileo, G.; Limongelli, G.; Gagliardi, M.G.; Santoro, G.; Digilio, M.C.; Di Salvo, G.; Ardorisio, R.; Miele, T.; Calabrò, R. A standard echocardiographic and tissue Doppler study of morphological and functional findings in children with hypertrophic cardiomyopathy compared to those with left ventricular hypertrophy in the setting of Noonan and LEOPARD syndromes. Cardiol. Young 2008, 18, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Lioncino, M.; Monda, E.; Verrillo, F.; Moscarella, E.; Calcagni, G.; Drago, F.; Marino, B.; Digilio, M.C.; Putotto, C.; Calabrò, P.; et al. Hypertrophic Cardiomyopathy in RASopathies: Diagnosis, Clinical Characteristics, Prognostic Implications, and Management. Heart Fail. Clin. 2022, 18, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Marino, B.; Digilio, M.C.; Toscano, A.; Giannotti, A.; Dallapiccola, B. Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J. Pediatr. 1999, 135, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Delogu, A.B.; Blandino, R.; Leoni, C.; Tartaglia, M.; Zampino, G. RASopathies and sigmoid-shaped ventricular septum morphology: Evidence of a previously unappreciated cardiac phenotype. Pediatr. Res. 2023, 93, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, H.; Ahrens-Nicklas, R.C.; Calderon-Anyosa, R.J.C.; Ritter, A.L.; Lin, K.Y.; Rossano, J.W.; Quartermain, M.D.; Banerjee, A. Genotype-phenotype association by echocardiography offers incremental value in patients with Noonan Syndrome with Multiple Lentigines. Pediatr. Res. 2021, 90, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.; Charron, P.; Gimeno-Blanes, J.; Helio, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Hennekam, R.C. Costello syndrome: An overview. Am. J. Med. Genet. Part C Semin. Med. Genet. 2003, 117, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.E.; Alexander, M.E.; Colan, S.D.; Kerr, B.; Rauen, K.A.; Noonan, J.; Baffa, J.; Hopkins, E.; Sol-Church, K.; Limongelli, G.; et al. Clinical, pathological, and molecular analyses of cardiovascular abnormalities in Costello syndrome: A Ras/MAPK pathway syndrome. Am. J. Med. Genet. Part A 2011, 155, 486–507. [Google Scholar] [CrossRef]
- Limongelli, G.; Monda, E.; Tramonte, S.; Gragnano, F.; Masarone, D.; Frisso, G.; Esposito, A.; Gravino, R.; Ammendola, E.; Salerno, G.; et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 299, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Pacileo, G.; Calabro, R. Is sudden cardiac death predictable in LEOPARD syndrome? Cardiol. Young 2006, 16, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Norrish, G.; Kolt, G.; Cervi, E.; Field, E.; Dady, K.; Ziółkowska, L.; Olivotto, I.; Favilli, S.; Passantino, S.; Limongelli, G.; et al. Clinical presentation and long-term outcomes of infantile hypertrophic cardiomyopathy: A European multicentre study. ESC Heart Fail. 2021, 8, 5057–5067. [Google Scholar] [CrossRef] [PubMed]
- Lynch, A.; Tatangelo, M.; Ahuja, S.; Steve Fan, C.P.; Min, S.; Lafreniere-Roula, M.; Papaz, T.; Zhou, V.; Armstrong, K.; Aziz, P.F.; et al. Risk of Sudden Death in Patients with RASopathy Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2023, 81, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Miron, A.; Lafreniere-Roula, M.; Steve Fan, C.P.; Armstrong, K.R.; Dragulescu, A.; Papaz, T.; Manlhiot, C.; Kaufman, B.; Butts, R.J.; Gardin, L.; et al. A Validated Model for Sudden Cardiac Death Risk Prediction in Pediatric Hypertrophic Cardiomyopathy. Circulation 2020, 142, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Boleti, O.D.; Roussos, S.; Norrish, G.; Field, E.; Oates, S.; Tollit, J.; Nepali, G.; Bhole, V.; Uzun, O.; Daubeney, P.E.F.; et al. Sudden cardiac death in childhood RASopathy-associated hypertrophic cardiomyopathy: Validation of the HCM risk-kids model and predictors of events. Int. J. Cardiol. 2023, 393, 131405. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J.; Miller, K.; Shaddy, R.E.; Lin, K.Y.; Hanna, B.D.; Ravishankar, C.; Rossano, J.W. Disopyramide use in infants and children with hypertrophic cardiomyopathy. Cardiol. Young 2018, 28, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Poterucha, J.T.; Johnson, J.N.; O’Leary, P.W.; Connolly, H.M.; Niaz, T.; Maleszewski, J.J.; Ackerman, M.J.; Cetta, F.; Dearani, J.A.; Eidem, B.W. Surgical Ventricular Septal Myectomy for Patients with Noonan Syndrome and Symptomatic Left Ventricular Outflow Tract Obstruction. Am. J. Cardiol. 2015, 116, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Gajarski, R.; Naftel, D.C.; Pahl, E.; Alejos, J.; Pearce, F.B.; Kirklin, J.K.; Zamberlan, M.; Dipchand, A.I.; Pediatric Heart Transplant Study, I. Outcomes of pediatric patients with hypertrophic cardiomyopathy listed for transplant. J. Heart Lung Transplant. 2009, 28, 1329–1334. [Google Scholar] [CrossRef]
- Artoni, A.; Selicorni, A.; Passamonti, S.M.; Lecchi, A.; Bucciarelli, P.; Cerutti, M.; Cianci, P.; Gianniello, F.; Martinelli, I. Hemostatic abnormalities in Noonan syndrome. Pediatrics 2014, 133, e1299–e1304. [Google Scholar] [CrossRef] [PubMed]
- Di Candia, F.; Marchetti, V.; Cirillo, F.; Di Minno, A.; Rosano, C.; Pagano, S.; Siano, M.A.; Falco, M.; Assunto, A.; Boccia, G.; et al. RASopathies and hemostatic abnormalities: Key role of platelet dysfunction. Orphanet J. Rare Dis. 2021, 16, 499. [Google Scholar] [CrossRef] [PubMed]
- Hoffner, B.; Benchich, K. Trametinib: A Targeted Therapy in Metastatic Melanoma. J. Adv. Pract. Oncol. 2018, 9, 741–745. [Google Scholar] [PubMed]
- Andelfinger, G.; Marquis, C.; Raboisson, M.J.; Theoret, Y.; Waldmuller, S.; Wiegand, G.; Gelb, B.D.; Zenker, M.; Delrue, M.A.; Hofbeck, M. Hypertrophic Cardiomyopathy in Noonan Syndrome Treated by MEK-Inhibition. J. Am. Coll. Cardiol. 2019, 73, 2237–2239. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Carli, D.; Giorgio, E.; Villar, A.M.; Cardaropoli, S.; Carbonara, C.; Campagnoli, M.F.; Galletto, P.; Palumbo, M.; Olivieri, S.; et al. MEK Inhibition in a Newborn with RAF1-Associated Noonan Syndrome Ameliorates Hypertrophic Cardiomyopathy but Is Insufficient to Revert Pulmonary Vascular Disease. Genes 2021, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Marin, T.M.; Keith, K.; Davies, B.; Conner, D.A.; Guha, P.; Kalaitzidis, D.; Wu, X.; Lauriol, J.; Wang, B.; Bauer, M.; et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J. Clin. Investig. 2011, 121, 1026–1043. [Google Scholar] [CrossRef] [PubMed]
- Gelb, B.D.; Yohe, M.E.; Wolf, C.; Andelfinger, G. New prospectives on treatment opportunities in RASopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 541–560. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Type of Defects | Congenital Heart Defects |
|---|---|
| Right heart obstructive lesions | Pulmonary valve stenosis |
| Supravalvular pulmonary stenosis | |
| Shunt lesions | Atrial septal defect |
| Ventricular septal defect | |
| Complete AV canal defect | |
| Partial AV canal defect | |
| Patent ductus arteriosus | |
| Left heart obstructive lesions | Aortic valve anomalies |
| Aortic coarctation | |
| Hypoplastic left heart syndrome | |
| Complex cyanotic lesions | Tetralogy of Fallot |
| Other | Mitral valve anomalies |
| Coronary artery anomalies |
| Gene | Cardiac Phenotype | Pathway |
|---|---|---|
| PTPN11 | PVS, HCM | Ras/Raf/MEK/MAPK, AKT/mTOR, FGF/BMP, TGF-β/BMP, WnT/β-catenin |
| HRAS | HCM | Ras/Raf/MEK/MAPK, Calcineurin/NFAT |
| KRAS | HRAS | Ras/Raf/MEK/MAPK, |
| NRAS | PVS, septal defects | Ras/Raf/MEK/MAPK, |
| RAF1 | HCM | Ras/Raf/MEK/MAPK, |
| RIT1 | HCM | Ras/Raf/MEK/MAPK, |
| SOS1 | PVS, HCM, septal defects | Ras/Raf/MEK/MAPK, |
| BRAF | PVS, HCM, septal defects | Ras/Raf/MEK/MAPK, |
| SHOC2 | HCM | Ras/Raf/MEK/MAPK, |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faienza, M.F.; Meliota, G.; Mentino, D.; Ficarella, R.; Gentile, M.; Vairo, U.; D’amato, G. Cardiac Phenotype and Gene Mutations in RASopathies. Genes 2024, 15, 1015. https://doi.org/10.3390/genes15081015
Faienza MF, Meliota G, Mentino D, Ficarella R, Gentile M, Vairo U, D’amato G. Cardiac Phenotype and Gene Mutations in RASopathies. Genes. 2024; 15(8):1015. https://doi.org/10.3390/genes15081015
Chicago/Turabian StyleFaienza, Maria Felicia, Giovanni Meliota, Donatella Mentino, Romina Ficarella, Mattia Gentile, Ugo Vairo, and Gabriele D’amato. 2024. "Cardiac Phenotype and Gene Mutations in RASopathies" Genes 15, no. 8: 1015. https://doi.org/10.3390/genes15081015
APA StyleFaienza, M. F., Meliota, G., Mentino, D., Ficarella, R., Gentile, M., Vairo, U., & D’amato, G. (2024). Cardiac Phenotype and Gene Mutations in RASopathies. Genes, 15(8), 1015. https://doi.org/10.3390/genes15081015