

Mild Phenotypes of Gyrate Atrophy in a Heterozygous Carrier with One Variant Allele of OAT
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Clinical Examinations, Biochemical Analysis, and Treatments
2.3. Genetic Testing and Pathogenicity Assessment
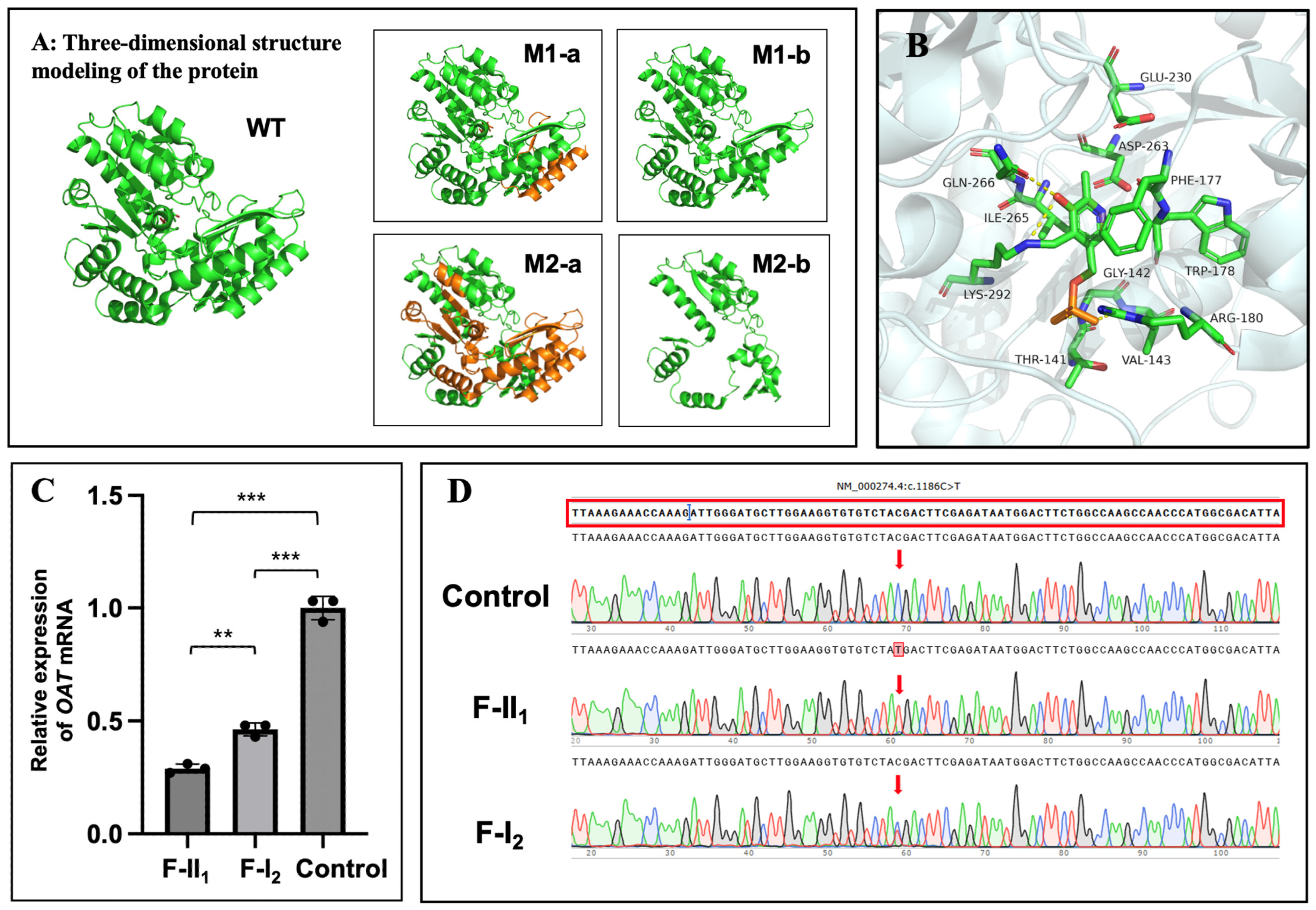
2.4. In Silico Analysis and Protein Structure Modelling
2.5. OAT mRNA Analysis
2.6. Statistical Analyses
3. Results
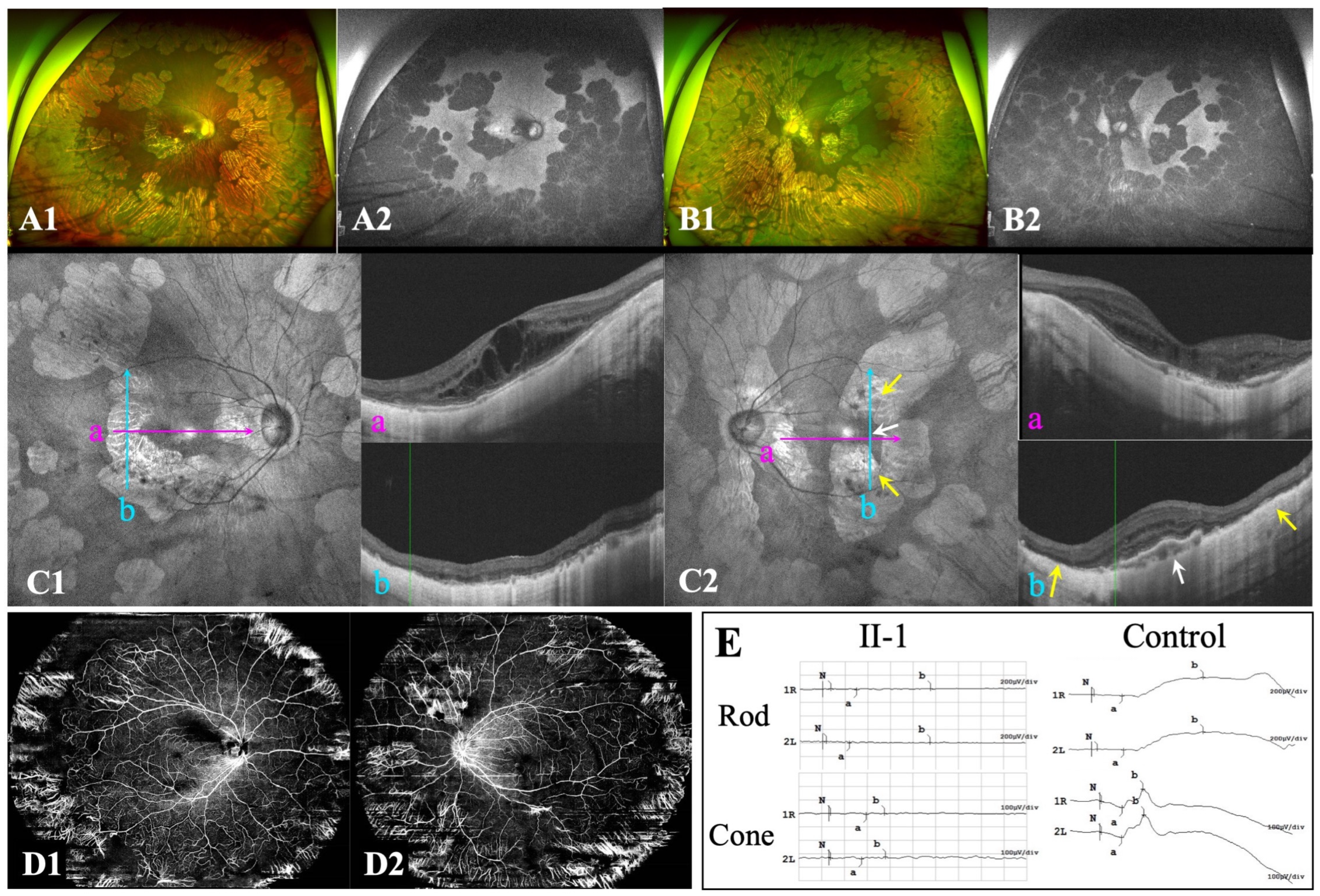
3.1. Ocular Characteristics
3.2. Hematologic Biochemical Findings
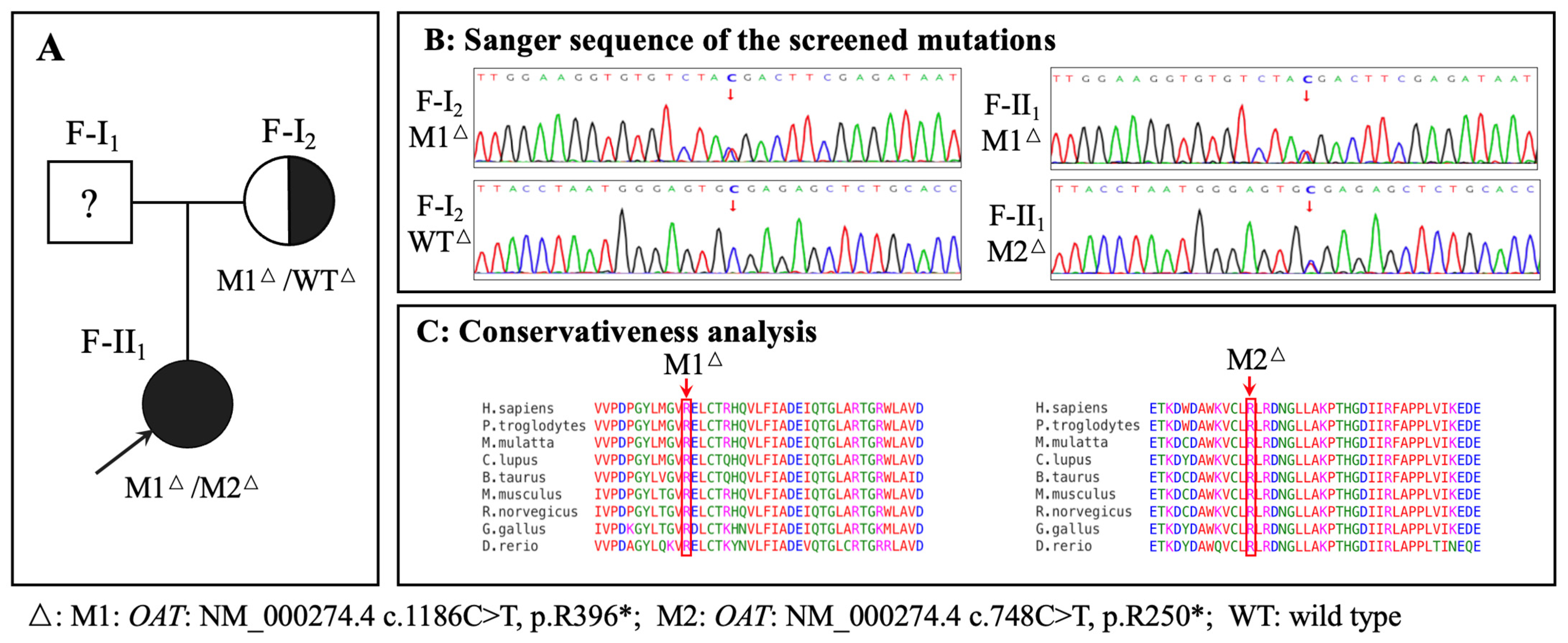
3.3. Genetic Findings
3.4. OAT mRNA Detection
3.5. Treatment and Follow-Up
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Inborn errors of metabolism: Gyrate atrophy. Adv. Exp. Med. Biol. 2018, 1085, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Montioli, R.; Bellezza, I.; Desbats, M.A.; Borri Voltattorni, C.; Salviati, L.; Cellini, B. Deficit of human ornithine aminotransferase in gyrate atrophy: Molecular, cellular, and clinical aspects. Biochim. Biophys. Acta. Proteins Proteom. 2021, 1869, 140555. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, V.; Benoit, L.A.; Crawford, P.; Harvey, P.T.; Shows, T.B.; Shih, V.E.; Gusella, J.F. The ornithine aminotransferase (OAT) locus: Analysis of RFLPs in gyrate atrophy. Am. J. Hum. Genet. 1988, 42, 365–372. [Google Scholar] [PubMed]
- Kaiser-Kupfer, M.I.; Valle, D.; Del Valle, L.A. A specific enzyme defect in gyrate atrophy. Am. J. Ophthalmol. 1978, 85, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Hayasaka, S.; Kodama, T.; Ohira, A. Retinal risks of high-dose ornithine supplements: A review. Br. J. Nutr. 2011, 106, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Peltola, K.E.; Näntö-Salonen, K.; Heinonen, O.J.; Jääskeläinen, S.; Heinänen, K.; Simell, O.; Nikoskelainen, E. Ophthalmologic heterogeneity in subjects with gyrate atrophy of choroid and retina harboring the L402P mutation of ornithine aminotransferase. Ophthalmology 2001, 108, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Gekka, T.; Hayashi, T.; Ida, H.; Ohashi, T.; Eto, Y.; Tsuneoka, H. OAT mutations and clinical features in two Japanese brothers with gyrate atrophy of the choroid and retina. Doc. Ophthalmol. 2014, 128, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.M.; Elnahry, A.G.; Tripathy, K.; Foster, R.E.; Mehanna, C.J.; Vishal, R.; Çavdarlı, C.; Arrigo, A.; Parodi, M.B. Analysis of optical coherence angiography in cystoid macular oedema associated with gyrate atrophy. Eye 2021, 35, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, K.; Chawla, R.; Sharma, Y.R.; Gogia, V. Ultrawide field fluorescein angiogram in a family with gyrate atrophy and foveoschisis. Oman J. Ophthalmol. 2016, 9, 104–106. [Google Scholar] [CrossRef]
- Kaiser-Kupfer, M.I.; de Monasterio, F.; Valle, D.; Walser, M.; Brusilow, S. Visual results of a long-term trial of a low-arginine diet in gyrate atrophy of choroid and retina. Ophthalmology 1981, 88, 307–310. [Google Scholar] [CrossRef]
- McInnes, R.R.; Arshinoff, S.A.; Bell, L.; McCulloch, C. Treatment of gyrate atrophy of the choroid and retina with low arginine diet. Trans. Am. Ophthalmol. Soc. 1980, 78, 226–242. [Google Scholar] [PubMed]
- Bergen, A.A.; Buijs, M.J.; Ten Asbroek, A.L.; Balfoort, B.M.; Boon, C.J.; Dutch GACR “Bird’s Eye View” Consortium; Brands, M.M.; Wanders, R.J.; van Karnebeek, C.D.; Houtkooper, R.H. Vision on gyrate atrophy: Why treat the eye? EMBO Mol. Med. 2024, 16, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Hames, A.; Khan, S.; Gilliland, C.; Goldman, L.; Lo, H.W.; Magda, K.; Keathley, J. Carriers of autosomal recessive conditions: Are they really “unaffected?”. J. Med. Genet. 2023, 61, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hillert, A.; Anikster, Y.; Belanger-Quintana, A.; Burlina, A.; Burton, B.K.; Carducci, C.; Chiesa, A.E.; Christodoulou, J.; Đorđević, M.; Desviat, L.R.; et al. The genetic landscape and epidemiology of phenylketonuria. Am. J. Hum. Genet. 2020, 107, 234–250. [Google Scholar] [CrossRef] [PubMed]
- van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat. Rev. Dis. Primers 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Polgreen, P.M.; Comellas, A.P. Clinical phenotypes of cystic fibrosis carriers. Annu. Rev. Med. 2022, 73, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Wagenaar, M.; ter Rahe, B.; van Aarem, A.; Huygen, P.; Admiraal, R.; Bleeker-Wagemakers, E.; Pinckers, A.; Kimberling, W.; Cremers, C. Clinical findings in obligate carriers of type I Usher syndrome. Am. J. Med. Genet. 1995, 59, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Matsui, K. Proposed classification of posterior staphylomas based on analyses of eye shape by three-dimensional magnetic resonance imaging and wide-field fundus imaging. Ophthalmology 2014, 121, 1798–1809. [Google Scholar] [CrossRef] [PubMed]
- Curtin, B.J. The posterior staphyloma of pathologic myopia. Trans. Am. Ophthalmol. Soc. 1977, 75, 67–86. [Google Scholar]
- Ohno-Matsui, K.; Wu, P.C.; Yamashiro, K.; Vutipongsatorn, K.; Fang, Y.; Cheung, C.M.G.; Lai, T.Y.Y.; Ikuno, Y.; Cohen, S.Y.; Gaudric, A.; et al. IMI Pathologic Myopia. Investig. Ophthalmol. Vis. Sci. 2021, 62, 5. [Google Scholar] [CrossRef]
- Ju, Y.; Zhang, L.; Gao, F.; Zong, Y.; Chen, T.; Ruan, L.; Chang, Q.; Zhang, T.; Huang, X. Genetic characteristics and clinical manifestations of foveal hypoplasia in familial exudative vitreoretinopathy. Am. J. Ophthalmol. 2024, 262, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Liao, K.; Zhao, Y.; Long, Z.; Zhu, S.; Wu, J.; Xiao, M.; Zhou, J.; Zhang, S.; Li, L.; et al. A novel system for the detection of spontaneous abortion-causing aneuploidy and its erroneous chromosome origins through the combination of low-pass copy number variation sequencing and NGS-based STR tests. J. Clin. Med. 2023, 12, 1809. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ohnaka, M.; Okuda-Ashitaka, E.; Kaneko, S.; Ando, A.; Maeda, M.; Furuta, K.; Suzuki, M.; Takahashi, K.; Ito, S. Induction of arginase II mRNA by nitric oxide using an in vitro model of gyrate atrophy of choroid and retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Sergouniotis, P.I.; Davidson, A.E.; Lenassi, E.; Devery, S.R.; Moore, A.T.; Webster, A.R. Retinal structure, function, and molecular pathologic features in gyrate atrophy. Ophthalmology 2012, 119, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Tauqeer, Z.; Yonekawa, Y. Familial exudative vitreoretinopathy: Pathophysiology, diagnosis, and management. Asia-Pac. J. Ophthalmol. 2018, 7, 176–182. [Google Scholar] [CrossRef]
- Snead, M.P.; Yates, J.R. Clinical and molecular genetics of Stickler syndrome. J. Med. Genet. 1999, 36, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Araújo, J.R.; Tavares-Ferreira, J.; Estrela-Silva, S.; Rocha, P.; Brandão, E.; Faria, P.A.; Falcão-Reis, F.; Rocha-Sousa, A. Wagner syndrome: Anatomic, functional and genetic characterization of a Portuguese family. Graefes Arch. Clin. Exp. Ophthalmol. 2018, 256, 163–171. [Google Scholar] [CrossRef]
- Ohno-Matsui, K.; Jonas, J.B. Posterior staphyloma in pathologic myopia. Prog. Retin. Eye Res. 2019, 70, 99–109. [Google Scholar] [CrossRef]
- Komori, S.; Ueno, S.; Ito, Y.; Sayo, A.; Meinert, M.; Kominami, T.; Inooka, D.; Kitagawa, M.; Nishida, K.; Takahashi, K.; et al. Steeper macular curvature in eyes with non-highly myopic retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3135–3141. [Google Scholar] [CrossRef] [PubMed]
- El Matri, L.; Falfoul, Y.; El Matri, K.; El Euch, I.; Ghali, H.; Habibi, I.; Hassairi, A.; Chaker, N.; Schorderet, D.; Chebil, A. Posterior staphylomas in non-highly myopic eyes with retinitis pigmentosa. Int. Ophthalmol. 2020, 40, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, S.; Max, E.E.; Kang, E.S. Phenylalanine hydroxylase activity in liver biopsies from hyperphenylalaninemia heterozygotes: Deviation from proportionality with gene dosage. Pediatr. Res. 1975, 9, 632–634. [Google Scholar] [CrossRef] [PubMed]
- Berry, H.K.; Hsieh, M.H.; Bofinger, M.K.; Schubert, W.K. Diagnosis of phenylalanine hydroxylase deficiency (phenylketonuria). Am. J. Dis. Child. 1982, 136, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.C.; Comellas, A.P.; Hornick, D.B.; Stoltz, D.A.; Cavanaugh, J.E.; Gerke, A.K.; Welsh, M.J.; Zabner, J.; Polgreen, P.M. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc. Natl. Acad. Sci. USA 2020, 117, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Keathley, J.; Garneau, V.; Zavala-Mora, D.; Heister, R.R.; Gauthier, E.; Morin-Bernier, J.; Green, R.; Vohl, M.C. A systematic review and recommendations around frameworks for evaluating scientific validity in nutritional genomics. Front. Nutr. 2021, 8, 789215. [Google Scholar] [CrossRef]
- Karousis, E.D.; Mühlemann, O. Nonsense-mediated mRNA decay begins where translation ends. Cold Spring Harb. Perspect. Biol. 2019, 11, a032862. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F-II1 | F-I2 | ||||
|---|---|---|---|---|---|
| Age (Years) | 19 | 54 | |||
| Chief complaint | Decreased vision bilaterally | High myopia and fluttering dark shadows in the right eye | |||
| Ophthalmic parameters | OD | OS | OD | OS | |
| IOP (mmHg) | 12.6 | 12.4 | 14.5 | 13.6 | |
| AL (mm) | 29.40 | 29.40 | 26.45 | 24.52 | |
| SE→BCVA | −17.00 D→20/66 | −18.25 D→20/66 | −9.00 D→20/28 | −2.75 D→20/28 | |
| Ocular manifestations | Punctate posterior subcapsular cataract | / | Nuclear cataract | Nuclear cataract | |
| Bilateral high myopia Bilateral PS (Type 1) Bilateral sharply demarcated circular areas of peripheral chorioretinal atrophy Bilateral macular atrophy | High myopia PS (Type 1) Focal circular areas of retinal atrophy / | Mild myopia Inferior PS (Type 5) / / | |||
| Haematological testing (μM) | Serum value | Serum value | Reference | ||
| Ornithine | 257.92 ↑ | 102.08 ↑ | 10–100 | ||
| Creatine | 94.83 ↓ | 121.75 | 95–1000 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, Y.; Zong, Y.; Li, X.; Gao, F.; Chang, Q.; Huang, X. Mild Phenotypes of Gyrate Atrophy in a Heterozygous Carrier with One Variant Allele of OAT. Genes 2024, 15, 1020. https://doi.org/10.3390/genes15081020
Ju Y, Zong Y, Li X, Gao F, Chang Q, Huang X. Mild Phenotypes of Gyrate Atrophy in a Heterozygous Carrier with One Variant Allele of OAT. Genes. 2024; 15(8):1020. https://doi.org/10.3390/genes15081020
Chicago/Turabian StyleJu, Yuqiao, Yuan Zong, Xiao Li, Fengjuan Gao, Qing Chang, and Xin Huang. 2024. "Mild Phenotypes of Gyrate Atrophy in a Heterozygous Carrier with One Variant Allele of OAT" Genes 15, no. 8: 1020. https://doi.org/10.3390/genes15081020