
Combined Analysis of Multi-Study miRNA and mRNA Expression Data Shows Overlap of Selected miRNAs Involved in West Nile Virus Infections

 ,
, 
Abstract
1. Introduction
1.1. Studying West Nile Virus Infections on Multiple Omics Levels
1.2. MicroRNAs and Their Effect on Target mRNAs
1.3. Using Gene-Set Tests on Target mRNAs to Study miRNA Expression Changes
2. Materials and Methods
2.1. Selected miRNA and mRNA Datasets
2.2. Differential Expression Analysis
2.3. Target Set Enrichment Analysis
3. Results
3.1. Differentially Expressed miRNAs Selected from miRNA Expression Data
3.2. Running Tests for miRNA Using Target Enrichment Analysis and Differential Expression Analysis
3.3. Differentially Expressed miRNAs Selected from mRNA Target Set Testing
3.4. Overlap of Findings from miRNA Analysis and Target Set Testing
3.5. Robustness Analysis of miRNAs Selected by Target Set Testing
3.6. Differential Expression Analysis on the mRNA Level
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ziegler, U.; Santos, P.D.; Groschup, M.H.; Hattendorf, C.; Eiden, M.; Höper, D.; Eisermann, P.; Keller, M.; Michel, F.; Klopfleisch, R. West Nile virus epidemic in Germany triggered by epizootic emergence, 2019. Viruses 2020, 12, 448. [Google Scholar] [CrossRef] [PubMed]
- Calistri, P.; Giovannini, A.; Hubalek, Z.; Ionescu, A.; Monaco, F.; Savini, G.; Lelli, R. Epidemiology of West Nile in Europe and in the Mediterranean basin. Open Virol. J. 2010, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R.; Hayes, E.B. West Nile virus in the Americas. Med. Clin. North Am. 2008, 92, 1307–1322. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control Epidemiological Update: West Nile Virus Transmission Season in Europe. 2023. Available online: https://www.ecdc.europa.eu/en/news-events/epidemiological-update-west-nile-virus-transmission-season-europe-2023-0 (accessed on 15 February 2024).
- Frost, M.J.; Zhang, J.; Edmonds, J.H.; Prow, N.A.; Gu, X.; Davis, R.; Hornitzky, C.; Arzey, K.E.; Finlaison, D.; Hick, P. Characterization of virulent west nile virus kunjin strain, australia, 2011. Emerg. Infect. Dis. 2012, 18, 792. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.; Popovici, F.; Cernescu, C.; Campbell, G.; Nedelcu, N. West Nile encephalitis epidemic in southeastern Romania. Lancet 1998, 352, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Marini, G.; Rosá, R.; Pugliese, A.; Heesterbeek, H. Exploring vector-borne infection ecology in multi-host communities: A case study of West Nile virus. J. Theor. Biol. 2017, 415, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.L.; Marfin, A.A.; Lanciotti, R.S.; Gubler, D.J. West nile virus. Lancet Infect. Dis. 2002, 2, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Ceauşu, E.; Erşcoiu, S.; Calistru, P.; Ispas, D.; Dorobăţ, O.; Homoş, M.; Bărbulescu, C.; Cojocaru, I.; Simion, C.; Cristea, C. Clinical manifestations in the West Nile virus outbreak. Rom. J. Virol. 1997, 48, 3–11. [Google Scholar]
- Sejvar, J.J.; Haddad, M.B.; Tierney, B.C.; Campbell, G.L.; Marfin, A.A.; Van Gerpen, J.A.; Fleischauer, A.; Leis, A.A.; Stokic, D.S.; Petersen, L.R. Neurologic manifestations and outcome of West Nile virus infection. JAMA 2003, 290, 511–515. [Google Scholar] [CrossRef]
- Kosch, R.; Delarocque, J.; Claus, P.; Becker, S.C.; Jung, K. Gene expression profiles in neurological tissues during West Nile virus infection: A critical meta-analysis. BMC Genom. 2018, 19, 530. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [PubMed]
- Hemandhar Kumar, S.; Tapken, I.; Kuhn, D.; Claus, P.; Jung, K. bootGSEA: A bootstrap and rank aggregation pipeline for multi-study and multi-omics enrichment analyses. Front. Bioinform. 2024, 4, 1380928. [Google Scholar] [CrossRef] [PubMed]
- Kreitmaier, P.; Katsoula, G.; Zeggini, E. Insights from multi-omics integration in complex disease primary tissues. Trends Genet. 2023, 39, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Wang, S.; Zhong, H.; Yang, Q.; Zhang, F.; Zhuang, Z.; Yuan, J.; Nie, X.; Wang, S. Integrative analyses reveal transcriptome-proteome correlation in biological pathways and secondary metabolism clusters in A. flavus in response to temperature. Sci. Rep. 2015, 5, 14582. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.M.; Mott, J.L. Overview of microRNA biology. Semin. Liver Dis. 2015, 35, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Love, T.M.; Moffett, H.F.; Novina, C.D. Not miR-ly small RNAs: Big potential for microRNAs in therapy. J. Allergy Clin. Immunol. 2008, 121, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.-u.; Miyazaki, H.; Ochiya, T. The roles of microRNAs in breast cancer. Cancers 2015, 7, 598–616. [Google Scholar] [CrossRef]
- Westholm, J.O.; Lai, E.C. Mirtrons: microRNA biogenesis via splicing. Biochimie 2011, 93, 1897–1904. [Google Scholar] [CrossRef]
- Abba, M.L.; Patil, N.; Leupold, J.H.; Moniuszko, M.; Utikal, J.; Niklinski, J.; Allgayer, H. MicroRNAs as novel targets and tools in cancer therapy. Cancer Lett. 2017, 387, 84–94. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Artmann, S.; Jung, K.; Bleckmann, A.; Beissbarth, T. Detection of simultaneous group effects in microRNA expression and related target gene sets. PLoS ONE 2012, 7, e38365. [Google Scholar] [CrossRef] [PubMed]
- Geistlinger, L.; Csaba, G.; Santarelli, M.; Ramos, M.; Schiffer, L.; Turaga, N.; Law, C.; Davis, S.; Carey, V.; Morgan, M. Toward a gold standard for benchmarking gene set enrichment analysis. Brief. Bioinform. 2021, 22, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Goeman, J.J.; Bühlmann, P. Analyzing gene expression data in terms of gene sets: Methodological issues. Bioinformatics 2007, 23, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Parkinson, H.; Kapushesky, M.; Shojatalab, M.; Abeygunawardena, N.; Coulson, R.; Farne, A.; Holloway, E.; Kolesnykov, N.; Lilja, P.; Lukk, M. ArrayExpress—A public database of microarray experiments and gene expression profiles. Nucleic Acids Res. 2007, 35, D747–D750. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The gene expression omnibus database. In Statistical Genomics: Methods and Protocols; Humana: New York, NY, USA, 2016; pp. 93–110. [Google Scholar]
- Suthar, M.S.; Brassil, M.M.; Blahnik, G.; McMillan, A.; Ramos, H.J.; Proll, S.C.; Belisle, S.E.; Katze, M.G.; Gale, M., Jr. A systems biology approach reveals that tissue tropism to West Nile virus is regulated by antiviral genes and innate immune cellular processes. PLoS Pathog. 2013, 9, e1003168. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Lancaster, A.; Wilkins, C.; Suthar, M.S.; Huang, A.; Vick, S.C.; Clepper, L.; Thackray, L.; Brassil, M.M.; Virgin, H.W. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Cho, H.; Proll, S.C.; Szretter, K.J.; Katze, M.G.; Gale, M., Jr.; Diamond, M.S. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 2013, 19, 458–464. [Google Scholar] [CrossRef]
- Clarke, P.; Leser, J.S.; Bowen, R.A.; Tyler, K.L. Virus-induced transcriptional changes in the brain include the differential expression of genes associated with interferon, apoptosis, interleukin 17 receptor A, and glutamate signaling as well as flavivirus-specific upregulation of tRNA synthetases. MBio 2014, 5, e00902-14. [Google Scholar] [CrossRef]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K. A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef]
- Feng, S.; Heath, E.; Jefferson, B.; Joslyn, C.; Kvinge, H.; Mitchell, H.D.; Praggastis, B.; Eisfeld, A.J.; Sims, A.C.; Thackray, L.B. Hypergraph models of biological networks to identify genes critical to pathogenic viral response. BMC Bioinform. 2021, 22, 287. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodological) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L. The multiMiR R package and database: Integration of microRNA–target interactions along with their disease and drug associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D. MicroRNA targets in Drosophila. Genome Biol. 2003, 4, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-D.; Lin, F.-M.; Wu, W.-Y.; Liang, C.; Huang, W.-C.; Chan, W.-L.; Tsai, W.-T.; Chen, G.-Z.; Lee, C.-J.; Chiu, C.-M. miRTarBase: A database curates experimentally validated microRNA–target interactions. Nucleic Acids Res. 2011, 39, D163–D169. [Google Scholar] [CrossRef] [PubMed]
- Sethupathy, P.; Corda, B.; Hatzigeorgiou, A.G. TarBase: A comprehensive database of experimentally supported animal microRNA targets. RNA 2006, 12, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA targeting efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA–target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Wu, D.; Lim, E.; Vaillant, F.; Asselin-Labat, M.-L.; Visvader, J.E.; Smyth, G.K. ROAST: Rotation gene set tests for complex microarray experiments. Bioinformatics 2010, 26, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Beissbarth, T.; Speed, T.P. GOstat: Find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 2004, 20, 1464–1465. [Google Scholar] [CrossRef]
- Majewski, I.J.; Ritchie, M.E.; Phipson, B.; Corbin, J.; Pakusch, M.; Ebert, A.; Busslinger, M.; Koseki, H.; Hu, Y.; Smyth, G.K. Opposing roles of polycomb repressive complexes in hematopoietic stem and progenitor cells. Blood J. Am. Soc. Hematol. 2010, 116, 731–739. [Google Scholar] [CrossRef]
- Kolde, R.; Laur, S.; Adler, P.; Vilo, J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 2012, 28, 573–580. [Google Scholar] [CrossRef]
- Kumar, M.; Nerurkar, V.R. Integrated Analysis of MicroRNAs and Their Disease Related Targets in the Brain of Mice Infected with West Nile Virus. Virology 2014, 452–453, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Natekar, J.P.; Rothan, H.A.; Arora, K.; Strate, P.G.; Kumar, M. Cellular microRNA-155 Regulates Virus-Induced Inflammatory Response and Protects against Lethal West Nile Virus Infection. Viruses 2020, 12, 9. [Google Scholar] [CrossRef]
- Cai, W.; Pan, Y.; Cheng, A.; Wang, M.; Yin, Z.; Jia, R. Regulatory Role of Host MicroRNAs in Flaviviruses Infection. Front. Microbiol. 2022, 13, 869441. [Google Scholar] [CrossRef]
- Maciejewski, H. Gene set analysis methods: Statistical models and methodological differences. Brief. Bioinform. 2014, 15, 504–518. [Google Scholar] [CrossRef]
- Bayerlová, M.; Jung, K.; Kramer, F.; Klemm, F.; Bleckmann, A.; Beißbarth, T. Comparative study on gene set and pathway topology-based enrichment methods. BMC Bioinform. 2015, 16, 334. [Google Scholar] [CrossRef] [PubMed]
- Martyniuk, C.J.; Martínez, R.; Kostyniuk, D.J.; Mennigen, J.A.; Zubcevic, J. Genetic ablation of bone marrow beta-adrenergic receptors in mice modulates miRNA-transcriptome networks of neuroinflammation in the paraventricular nucleus. Physiol. Genom. 2020, 52, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Ueta, M.; Nishigaki, H.; Komai, S.; Mizushima, K.; Tamagawa-Mineoka, R.; Naito, Y.; Katoh, N.; Sotozono, C.; Kinoshita, S. Positive Regulation of Innate Immune Response by MiRNA-Let-7a-5p. Front. Genet. 2022, 13, 1025539. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Ai, J.; Xie, Z.; Zhou, C.; Liu, C.; Zhang, H.; Shen, K. Dynamic expression of viral and cellular microRNAs in infectious mononucleosis caused by primary Epstein-Barr virus infection in children. Virol. J. 2015, 12, 208. [Google Scholar] [CrossRef] [PubMed]
- Carnino, J.M.; Lee, H.; Smith, L.C.; Sunil, V.R.; Rancourt, R.C.; Vayas, K.; Cervelli, J.; Kwok, Z.H.; Ni, K.; Laskin, J.D. Microvesicle-derived miRNAs regulate proinflammatory macrophage activation in the lung following ozone exposure. Toxicol. Sci. 2022, 187, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Akbar, I.; Kumari, B.; Vrati, S.; Basu, A.; Banerjee, A. Japanese Encephalitis Virus-Induced Let-7a/B Interacted with the NOTCH - TLR 7 Pathway in Microglia and Facilitated Neuronal Death via Caspase Activation. J. Neurochem. 2019, 149, 518–534. [Google Scholar] [CrossRef]
- Gamdzyk, M.; Doycheva, D.M.; Kang, R.; Tang, H.; Travis, Z.D.; Tang, J.; Zhang, J.H. GW0742 Activates MiR-17-5p and Inhibits TXNIP/NLRP3-Mediated Inflammation after Hypoxic-Ischaemic Injury in Rats and in PC12 Cells. J. Cell. Mol. Med. 2020, 24, 12318–12330. [Google Scholar] [CrossRef] [PubMed]
- Öksüz, Z.; Gragnani, L.; Lorini, S.; Temel, G.Ö.; Serin, M.S.; Zignego, A.L. Evaluation of Plasma miR-17-5p, miR-24-3p and miRNA-223-3p Profile of Hepatitis C Virus-Infected Patients after Treatment with Direct-Acting Antivirals. J. Pers. Med. 2023, 13, 1188. [Google Scholar] [CrossRef] [PubMed]
- Fayyad-Kazan, M.; Makki, R.; Skafi, N.; El Homsi, M.; Hamade, A.; El Majzoub, R.; Hamade, E.; Fayyad-Kazan, H.; Badran, B. Circulating miRNAs: Potential diagnostic role for coronavirus disease 2019 (COVID-19). Infect. Genet. Evol. 2021, 94, 105020. [Google Scholar] [CrossRef]
- Ge, Y.; Lin, D.; Cui, B.; Zhang, L.; Li, S.; Wang, Z.; Ma, J. Effects of long noncoding RNA H19 on isoflurane-induced cognitive dysregulation by promoting neuroinflammation. Neuroimmunomodulation 2022, 29, 117–127. [Google Scholar] [CrossRef]
- Kanokudom, S.; Vilaivan, T.; Wikan, N.; Thepparit, C.; Smith, D.R.; Assavalapsakul, W. MiR-21 Promotes Dengue Virus Serotype 2 Replication in HepG2 Cells. Antivir. Res. 2017, 142, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Zhao, K.; Lan, Y.; Li, Z.; Ding, N.; Su, J.; Lu, H.; Song, D.; Gao, F.; He, W. miR-21a-5p contributes to porcine hemagglutinating encephalomyelitis virus proliferation via targeting CASK-interactive protein1 in vivo and vitro. Front. Microbiol. 2017, 8, 304. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, Y.; Liang, T.; Hu, Y.; Tang, H.; Song, D.; Fang, H. The regulatory effect of microRNA-21a-3p on the promotion of telocyte angiogenesis mediated by PI3K (p110α)/AKT/mTOR in LPS induced mice ARDS. J. Transl. Med. 2019, 17, 427. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Huang, M.; Chen, L. Mechanism of MiR-132-3p Promoting Neuroinflammation and Dopaminergic Neurodegeneration in Parkinson’s Disease. Eneuro 2022, 9, ENEURO.0393-21.2021. [Google Scholar] [CrossRef]
- Qu, J.; Xiong, X.; Hujie, G.; Ren, J.; Yan, L.; Ma, L. MicroRNA-132-3p Alleviates Neuron Apoptosis and Impairments of Learning and Memory Abilities in Alzheimer’s Disease by Downregulation of HNRNPU Stabilized BACE1. Cell Cycle 2021, 20, 2309–2320. [Google Scholar] [CrossRef]
- Chen, W.; Ma, X.; Zhang, P.; Li, Q.; Liang, X.; Liu, J. MiR-212-3p Inhibits LPS-Induced Inflammatory Response through Targeting HMGB1 in Murine Macrophages. Exp. Cell Res. 2017, 350, 318–326. [Google Scholar] [CrossRef]
- Bampali, M.; Kouvela, A.; Kesesidis, N.; Kassela, K.; Dovrolis, N.; Karakasiliotis, I. West Nile Virus Subgenomic RNAs Modulate Gene Expression in a Neuronal Cell Line. Viruses 2024, 16, 812. [Google Scholar] [CrossRef]
- Gorman, M.J.; Poddar, S.; Farzan, M.; Diamond, M.S. The interferon-stimulated gene Ifitm3 restricts West Nile virus infection and pathogenesis. J. Virol. 2016, 90, 8212–8225. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Weidner, J.M.; Qing, M.; Pan, X.B.; Guo, H.; Xu, C.; Zhang, X.; Birk, A.; Chang, J.; Guo, J.T.; et al. Identification of five interferon-induced cellular proteins that inhibit west nile virus and dengue virus infections. J. Virol. 2010, 84, 8332–8341. [Google Scholar] [CrossRef]
- Yakub, I.; Lillibridge, K.M.; Moran, A.; Gonzalez, O.Y.; Belmont, J.; Gibbs, R.A.; Tweardy, D.J. Single nucleotide polymorphisms in genes for 2′-5′-oligoadenylate synthetase and RNase L in patients hospitalized with West Nile virus infection. J. Infect. Dis. 2005, 192, 1741–1748. [Google Scholar] [CrossRef]
- Gurevitch, J.; Koricheva, J.; Nakagawa, S.; Stewart, G. Meta-analysis and the science of research synthesis. Nature 2018, 555, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Noble, D.W.; Senior, A.M.; Lagisz, M. Meta-evaluation of meta-analysis: Ten appraisal questions for biologists. BMC Biol. 2017, 15, 18. [Google Scholar] [CrossRef] [PubMed]
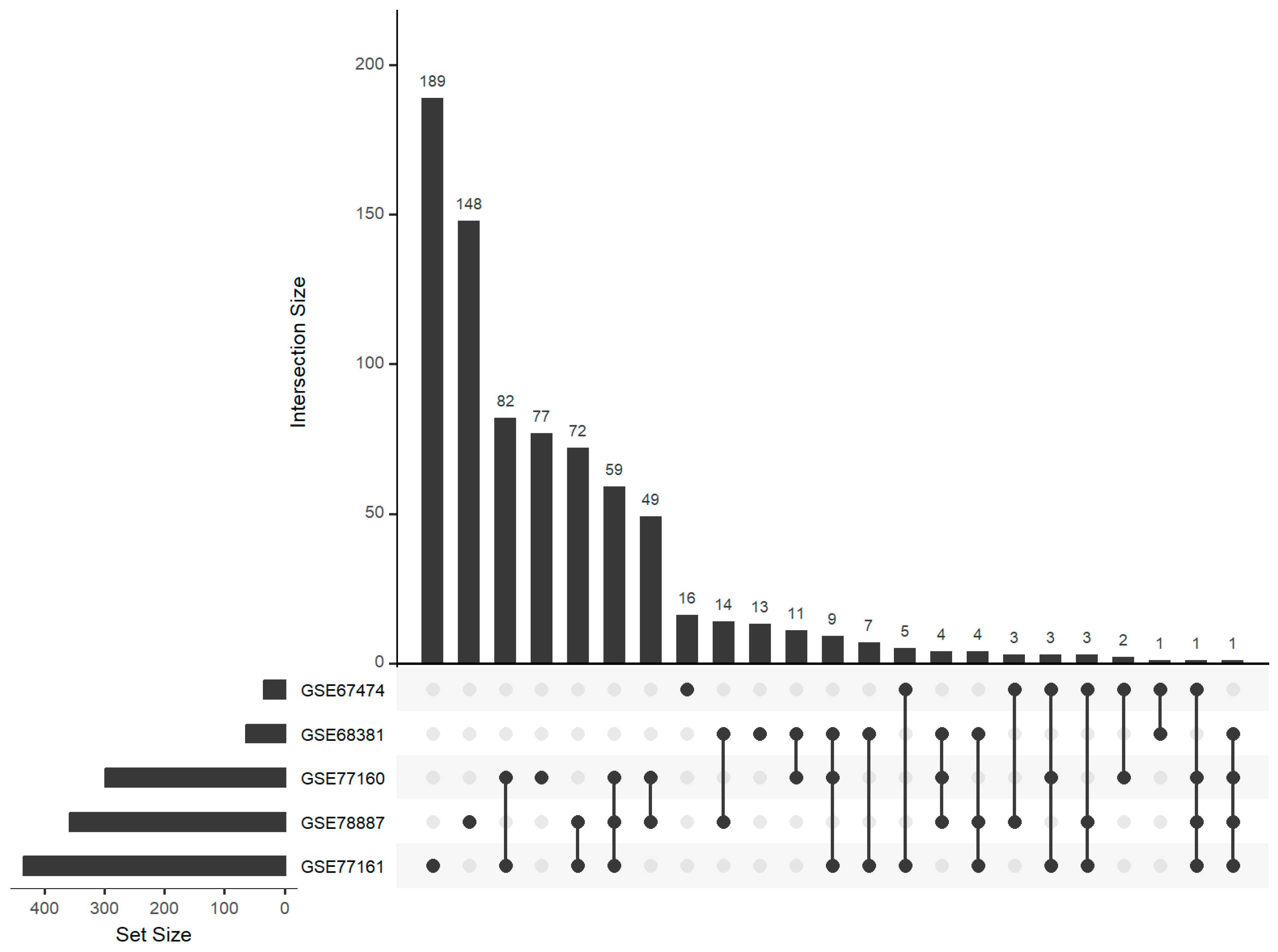

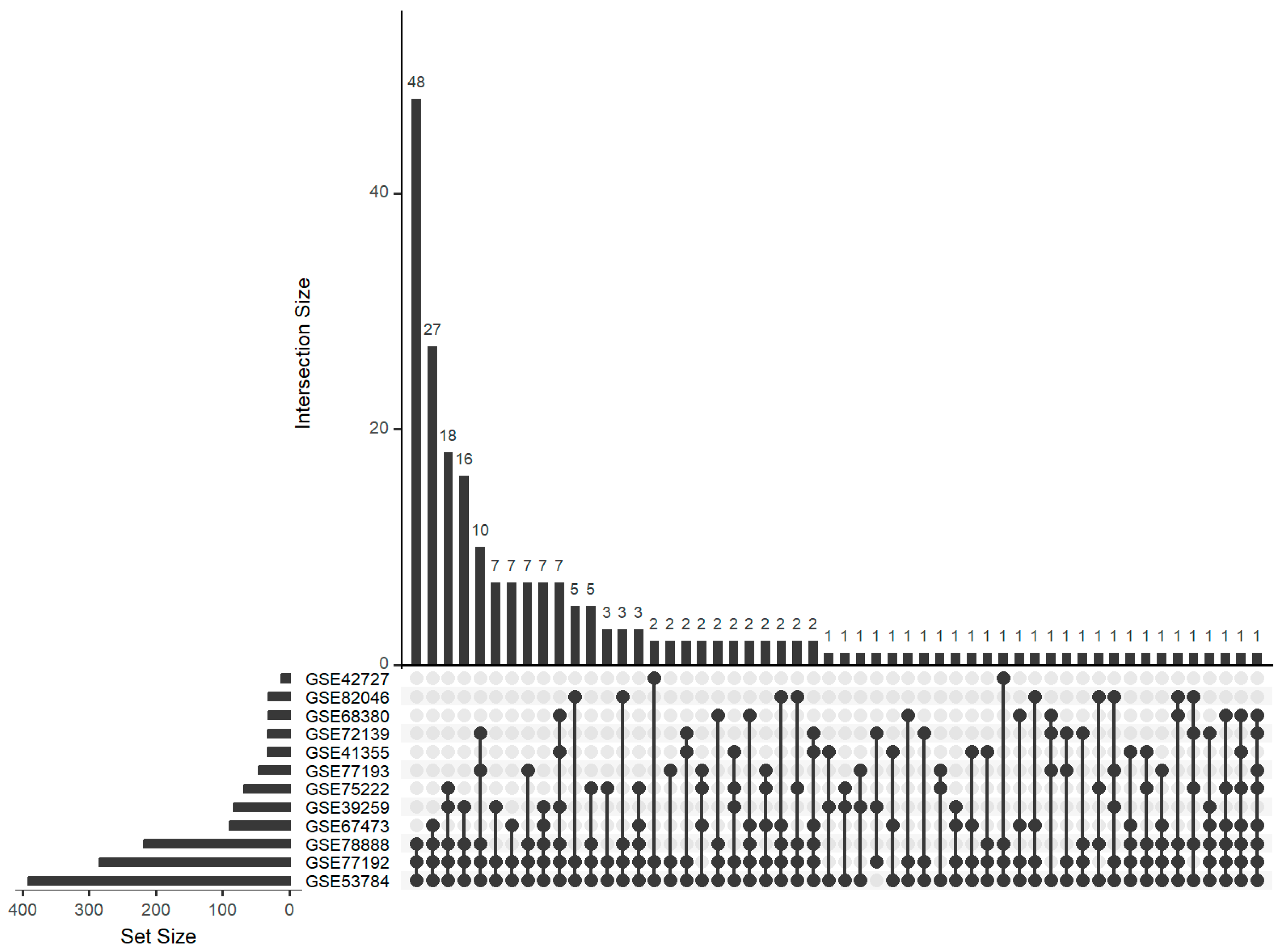

{kind=link}
{kind=link}
{kind=link}
| Databank ID | Mouse Line | Tissue | Time after Infection | Sample Size (Control + WNV) | Reference | Omics Level |
|---|---|---|---|---|---|---|
| GSE39259 | n.a. | Liver | 4 days | 2 + 3 | [28] | mRNA |
| GSE41355 | n.a. | Bone marrow-derived dendritic cells | 24 h | 3 + 3 | [29] | mRNA |
| GSE42727 | C57BL/6 | Cortical and granule neuron | 12 + 24 h | 10 + 10 | [30] | mRNA |
| GSE53784 | SW | Whole brain | n.a. | 3 + 3 | [31] | mRNA |
| GSE72139 | C57Bl/6 | Hippocampus | 25 days | 4 + 8 | [32] | mRNA |
| GSE74628 | C57BL/6 | Primary myeloid dendritic cell | 24 h | 2 + 3 | [33] | mRNA |
| GSE75222 | C57BL/6 | Primary myeloid dendritic cell | 24 h | 3 + 2 | [33] | mRNA |
| GSE82046 | crossline genetic background | Spleen | 28 days | 19 + 16 | n.a. | mRNA |
| Databank ID | Mouse Line | Tissue | Time after Infection | Sample Size (Control + WNV) | Reference | Omics Level |
|---|---|---|---|---|---|---|
| GSE67473 | C57Bl/6 | Cortical neuron | 24 h | 6 + 6 | [33] | mRNA |
| GSE67474 | C57Bl/6 | Cortical neuron | 12 h | 5 + 6 | [33] | miRNA |
| GSE68380 | C57Bl/6 | Granule cell neurons | 24 h | 6 + 6 | [33] | mRNA |
| GSE68381 | C57Bl/6 | Granule cell neurons | 12 h | 6 + 6 | [33] | miRNA |
| GSE77192 | C57Bl/6 | Cerebellum | 4 days | 5 + 5 | [33] | mRNA |
| GSE77160 | C57Bl/6 | Cerebellum | 4 days | 5 + 5 | [33] | miRNA |
| GSE77193 | C57BL/6 | Cortex | 4 days | 5 + 5 | [33] | mRNA |
| GSE77161 | C57BL/6 | Cortex | 4 days | 5 + 5 | [33] | miRNA |
| GSE78888 | C57BL/6 | Popliteal lymph node | 1 days | 3 + 5 | [33] | mRNA |
| GSE78887 | C57BL/6 | Popliteal lymph node | 2 days | 3 + 5 | [33] | miRNA |
| Databank ID | # miRNA in Dataset | # p < 0.05 | # pFDR < 0.05 | # |log2FC| > 1 |
|---|---|---|---|---|
| GSE78887 | 1247 | 359 | 199 | 53 |
| GSE77161 | 1247 | 435 | 119 | 13 |
| GSE77160 | 1247 | 298 | 59 | 5 |
| GSE68381 | 1247 | 64 | 1 | 2 |
| GSE67474 | 1247 | 34 | 2 | 0 |
| WNV | West Nile | Virus | Viral | HIV | Covid | Inflammation | Inflammatory | Encephalitis | Neuroinflammation | Hits | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| miR-17-5p | 0 | 0 | 13 | 10 | 1 | 5 | 13 | 5 | 0 | 1 | 48 |
| let-7a-5p | 0 | 0 | 5 | 3 | 1 | 3 | 3 | 8 | 0 | 1 | 24 |
| miR-15b-5p | 0 | 0 | 2 | 2 | 1 | 3 | 3 | 7 | 0 | 0 | 18 |
| miR-132-3p | 0 | 0 | 3 | 0 | 0 | 0 | 6 | 5 | 1 | 2 | 17 |
| miR-185-5p | 0 | 0 | 4 | 0 | 0 | 2 | 3 | 6 | 0 | 0 | 15 |
| miR-21a-5p | 0 | 0 | 1 | 1 | 0 | 0 | 7 | 3 | 0 | 2 | 14 |
| miR-381-3p | 0 | 0 | 3 | 0 | 1 | 0 | 6 | 1 | 2 | 0 | 13 |
| miR-92a-3p | 0 | 0 | 1 | 1 | 0 | 5 | 1 | 1 | 1 | 0 | 10 |
| miR-212-3p | 0 | 0 | 0 | 0 | 0 | 1 | 3 | 4 | 0 | 1 | 9 |
| miR-18a-5p | 0 | 0 | 1 | 0 | 1 | 2 | 3 | 1 | 0 | 0 | 8 |
| miR-363-3p | 0 | 0 | 2 | 1 | 0 | 0 | 1 | 2 | 0 | 0 | 6 |
| miR-665-3p | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 3 |
| miR-7a-5p | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 3 |
| Databank ID of mRNA Data | Databank ID of Parallel miRNA Data | # mRNA in Dataset | # p < 0.05 | # pFDR < 0.05 | # |log2FC| > 1 |
| GSE39259 | 21,200 | 827 | 0 | 0 | |
| GSE41355 | 18,120 | 3479 | 1510 | 211 | |
| GSE42727 | 21,609 | 544 | 0 | 0 | |
| GSE53784 | 24,214 | 9231 | 6409 | 781 | |
| GSE67473 | GSE67474 | 24,162 | 4048 | 1335 | 210 |
| GSE68380 | GSE68381 | 24,159 | 1067 | 140 | 78 |
| GSE72139 | 30,869 | 3786 | 534 | 346 | |
| GSE74628 | 24,162 | 6405 | 1552 | 719 | |
| GSE75222 | 24,162 | 3195 | 81 | 73 | |
| GSE77192 | GSE77160 | 24,162 | 14,981 | 13,618 | 4172 |
| GSE77193 | GSE77161 | 24,162 | 9356 | 4971 | 661 |
| GSE78888 | GSE78887 | 24,162 | 6812 | 2969 | 719 |
| GSE82046 | 23,736 | 1070 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Böge, F.L.; Ruff, S.; Hemandhar Kumar, S.; Selle, M.; Becker, S.; Jung, K. Combined Analysis of Multi-Study miRNA and mRNA Expression Data Shows Overlap of Selected miRNAs Involved in West Nile Virus Infections. Genes 2024, 15, 1030. https://doi.org/10.3390/genes15081030
Böge FL, Ruff S, Hemandhar Kumar S, Selle M, Becker S, Jung K. Combined Analysis of Multi-Study miRNA and mRNA Expression Data Shows Overlap of Selected miRNAs Involved in West Nile Virus Infections. Genes. 2024; 15(8):1030. https://doi.org/10.3390/genes15081030
Chicago/Turabian StyleBöge, Franz Leonard, Sergej Ruff, Shamini Hemandhar Kumar, Michael Selle, Stefanie Becker, and Klaus Jung. 2024. "Combined Analysis of Multi-Study miRNA and mRNA Expression Data Shows Overlap of Selected miRNAs Involved in West Nile Virus Infections" Genes 15, no. 8: 1030. https://doi.org/10.3390/genes15081030
APA StyleBöge, F. L., Ruff, S., Hemandhar Kumar, S., Selle, M., Becker, S., & Jung, K. (2024). Combined Analysis of Multi-Study miRNA and mRNA Expression Data Shows Overlap of Selected miRNAs Involved in West Nile Virus Infections. Genes, 15(8), 1030. https://doi.org/10.3390/genes15081030