
A Genotype/Phenotype Study of KDM5B-Associated Disorders Suggests a Pathogenic Effect of Dominantly Inherited Missense Variants
 , , , , , , , , , , and add
Show full author list
, , , , , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Methods
3. Results
3.1. Genotypic Findings in New Cases of KDM5B-Related Disorder
3.2. Clinical Findings in New Cases of KDM5B-Related Disorder
3.3. Comparison of Our Findings with the Literature
3.4. Comparison of Phenotypes Based on Variant Type
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Travers, A.; Muskhelishvili, G. DNA structure and function. FEBS J. 2015, 282, 2279–2295. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.D.; Poirier, M.G. Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev. 2015, 115, 2274–2295. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.R.; Lambert, S.A. Transcription factors read epigenetics. Science 2017, 356, 489–490. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.; Chen, T. Chapter 3—Writers, erasers, and readers of DNA and histone methylation marks. In Epigenetic Cancer Therapy, 2nd ed.; Gray, S.G., Ed.; Academic Press: Boston, MA, USA, 2023; pp. 39–63. [Google Scholar]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- McKusick-Nathans Institute of Genetic Medicine. Online Mendelian Inheritance in Man, OMIM®. 2022. Available online: https://omim.org/ (accessed on 5 December 2023).
- Vallianatos, C.N.; Iwase, S. Disrupted Intricacy of Histone H3K4 Methylation in Neurodevelopmental Disorders. Epigenomics 2015, 7, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, Y.; Wu, H.; Qiu, X.; Yu, X.; Wang, R.; Zhong, J.; Peng, J. De novo variants in PHF21A cause intellectual developmental disorder with behavioral abnormalities and craniofacial dysmorphism with or without seizures: A case report and literature review. Seizure 2023, 111, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Sobering, A.K.; Bryant, L.M.; Li, D.; McGaughran, J.; Maystadt, I.; Moortgat, S.; Graham, J.M.; van Haeringen, A.; Ruivenkamp, C.; Cuperus, R.; et al. Variants in PHF8 cause a spectrum of X-linked neurodevelopmental disorders and facial dysmorphology. Hum. Genet. Genom. Adv. 2022, 3, 100102. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, S.; Di Fede, E.; Bernardelli, C.; Lettieri, A.; Parodi, C.; Grazioli, P.; Colombo, E.A.; Ancona, S.; Milani, D.; Ottaviano, E.; et al. KMT2A: Umbrella Gene for Multiple Diseases. Genes 2022, 13, 514. [Google Scholar] [CrossRef] [PubMed]
- Õunap, K.; Puusepp-Benazzouz, H.; Peters, M.; Vaher, U.; Rein, R.; Proos, A.; Field, M.; Reimand, T. A novel c.2T>C mutation of the KDM5C/JARID1C gene in one large family with X-linked intellectual disability. Eur. J. Med. Genet. 2012, 55, 178–184. [Google Scholar] [CrossRef]
- Jensen, L.R.; Amende, M.; Gurok, U.; Moser, B.; Gimmel, V.; Tzschach, A.; Janecke, A.R.; Tariverdian, G.; Chelly, J.; Fryns, J.-P.; et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet. 2005, 76, 227–236. [Google Scholar] [CrossRef]
- Carmignac, V.; Nambot, S.; Lehalle, D.; Callier, P.; Moortgat, S.; Benoit, V.; Ghoumid, J.; Delobel, B.; Smol, T.; Thuillier, C.; et al. Further delineation of the female phenotype with KDM5C disease causing variants: 19 new individuals and review of the literature. Clin. Genet. 2020, 98, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011, 478, 57–63. [Google Scholar] [CrossRef] [PubMed]
- El Hayek, L.; Tuncay, I.O.; Nijem, N.; Russell, J.; Ludwig, S.; Kaur, K.; Li, X.; Anderton, P.; Tang, M.; Gerard, A.; et al. KDM5A mutations identified in autism spectrum disorder using forward genetics. Elife 2020, 9, e56883. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Tian, R.; Ge, T.; Lam, M.; Sanchez-Andrade, G.; Singh, T.; Urpa, L.; Liu, J.Z.; Sanderson, M.; Rowley, C.; et al. The impact of rare protein coding genetic variation on adult cognitive function. Nat. Genet. 2023, 55, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Faundes, V.; Newman, W.G.; Bernardini, L.; Canham, N.; Clayton-Smith, J.; Dallapiccola, B.; Davies, S.J.; Demos, M.K.; Goldman, A.; Gill, H.; et al. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am. J. Hum. Genet. 2018, 102, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.C.; Jones, W.D.; McIntyre, R.; Sanchez-Andrade, G.; Sanderson, M.; Stephenson, J.D.; Jones, C.P.; Handsaker, J.; Gallone, G.; Bruntraeger, M.; et al. Quantifying the contribution of recessive coding variation to developmental disorders. Science 2018, 362, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.; Thiruvahindrapuram, B.; Chan, A.J.; Engchuan, W.; Higginbotham, E.J.; Howe, J.L.; Loureiro, L.O.; Reuter, M.S.; Roshandel, D.; Whitney, J.; et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 2022, 185, 4409–4427.e18. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, T.V.; Williams, Z.P.; Kline, T.; Rajendran, S.; Augustine, F.; Wright, N.; Sullivan, C.A.W.; Olfson, E.; Abdallah, S.B.; Liu, W.; et al. Primary complex motor stereotypies are associated with de novo damaging DNA coding mutations that identify KDM5B as a risk gene. PLoS ONE 2023, 18, e0291978. [Google Scholar] [CrossRef] [PubMed]
- Sobreira, N.; Schiettecatte, F.; Valle, D.; Hamosh, A. GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015, 36, 928–930. [Google Scholar] [CrossRef]
- Lebrun, N.; Mehler-Jacob, C.; Poirier, K.; Zordan, C.; Lacombe, D.; Carion, N.; Billuart, P.; Bienvenu, T. Novel KDM5B splice variants identified in patients with developmental disorders: Functional consequences. Gene 2018, 679, 305–313. [Google Scholar] [CrossRef]
- Bell, S.; Rousseau, J.; Peng, H.; Aouabed, Z.; Priam, P.; Theroux, J.-F.; Jefri, M.; Tanti, A.; Wu, H.; Kolobova, I.; et al. Mutations in ACTL6B Cause Neurodevelopmental Deficits and Epilepsy and Lead to Loss of Dendrites in Human Neurons. Am. J. Hum. Genet. 2019, 104, 815–834. [Google Scholar] [CrossRef] [PubMed]
- Lebon, S.; Quinodoz, M.; Peter, V.G.; Gengler, C.; Blanchard, G.; Cina, V.; Campos-Xavier, B.; Rivolta, C.; Superti-Furga, A. Agenesis of the Corpus Callosum with Facial Dysmorphism and Intellectual Disability in Sibs Associated with Com-pound Heterozygous KDM5B Variants. Genes 2021, 12, 1397. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.J.; Piao, L.; Xi, Y.; Rincon-Arano, H.; Rothbart, S.B.; Peng, D.; Wen, H.; Larson, C.; Zhang, X.; Zheng, X.; et al. The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell Rep. 2014, 6, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.M.; Otero, M.G.; Grand, K.; Choi, A.; Graham, J.M.; Young, J.I.; Mackay, J.P. The NuRD complex and macrocephaly associated neurodevelopmental disorders. Am. J. Med. Genet. Part C Semin. Med. Genet. 2019, 181, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Lazar, H.P.; Kurolap, A.; Martinez, A.F.; Paperna, T.; Cohen, L.; Smeland, M.F.; Whalen, S.; Heide, S.; Keren, B.; et al. The CHD4-related syndrome: A comprehensive investigation of the clinical spectrum, genotype–phenotype correlations, and molecular basis. Genet. Med. 2020, 22, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Mangano, G.D.; Antona, V.; Calì, E.; Fontana, A.; Salpietro, V.; Houlden, H.; Veggiotti, P.; Nardello, R. A complex epileptic and dysmorphic phenotype associated with a novel frameshift KDM5B variant and deletion of SCN gene cluster. Seizure 2022, 97, 20–22. [Google Scholar] [CrossRef]
- Barton, A.R.; Hujoel, M.L.A.; Mukamel, R.E.; Sherman, M.A.; Loh, P.-R. A spectrum of recessiveness among Mendelian disease variants in UK Biobank. Am. J. Hum. Genet. 2022, 109, 1298–1307. [Google Scholar] [CrossRef]
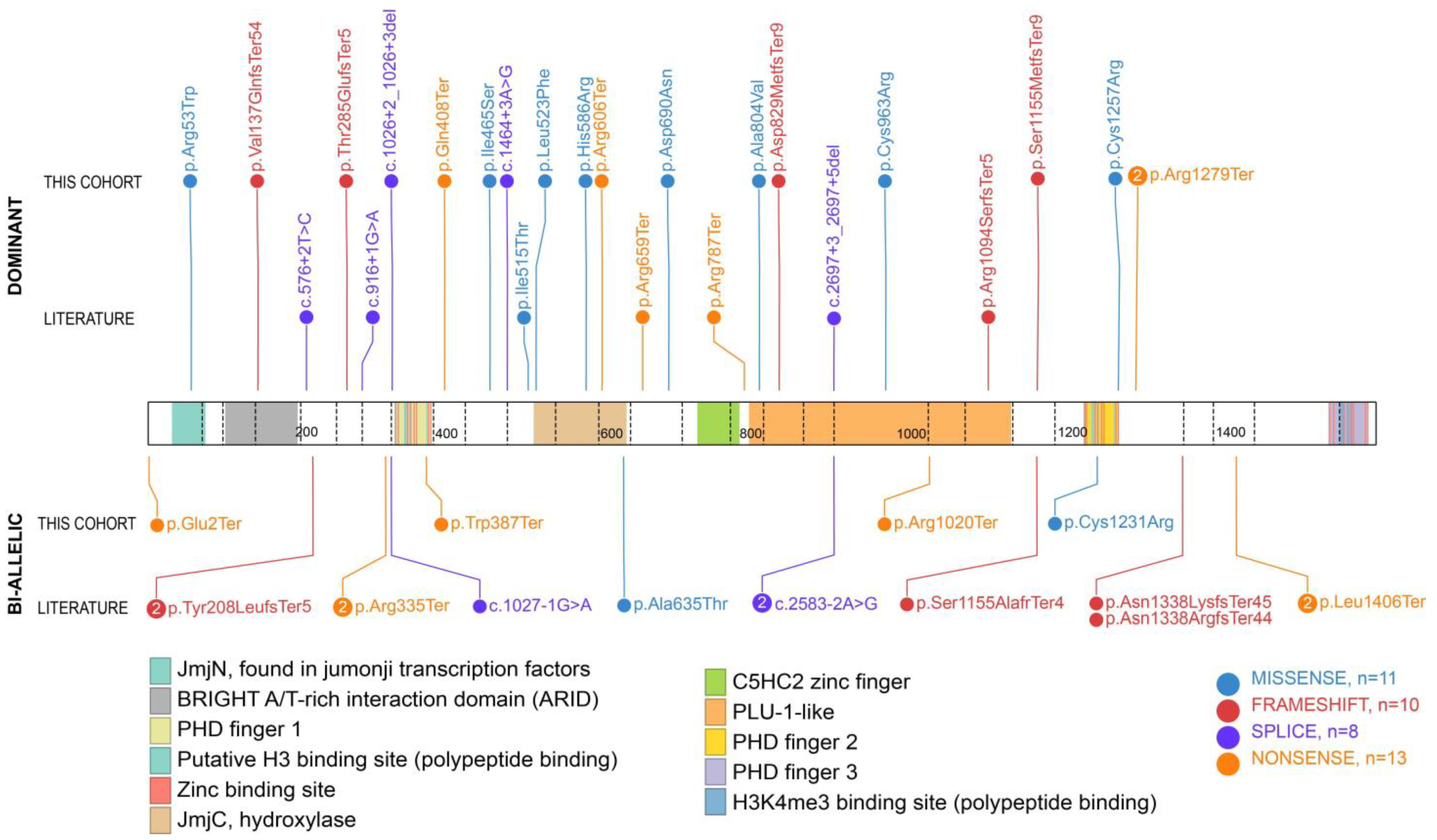


{kind=link}
{kind=link}
| No. | Genetic Variant (hg19/GRCh37) | cDNA Variant (NM_001314042.1) | Protein Variant | Inheritance | Predicted Effect | Pathogenicity Score (CADD) a | gnomAD (Allele Count) b |
|---|---|---|---|---|---|---|---|
| 1 | chr1:g.207207771A>G | c.3769T>C | p.Cys1257Arg | De novo | Missense | 30 | N/A |
| 2 | chr1:g.20716061A>G | c.2887T>C | p.Cys963Arg | De novo | Missense | 22.2 | N/A |
| 3 | chr1:g.202718129C>T | c.2068G>A | p.Asp690Asn | De novo | Missense | 25.8 | N/A |
| 4 | chr1:g.202722085T>C | c.1757A>G | p.His586Arg | De novo | Missense | 26 | N/A |
| 5 | chr1:g.202715006G>A | c.2411T>C | p.Ala804Val | De novo | Missense | 27.8 | N/A |
| 6 | chr1:g.202772777T>G | c.157C>T | p.Arg53Trp | De novo | Missense | 28.3 | N/A |
| 7 | chr1:g.202725556A>C | c.1394T>G | p.Ile465Ser | De novo | Missense | 29.1 | N/A |
| 8 | chr1:g.202724476C>G | c.1569C>G | p.Leu523Phe | De novo | Missense | 23.9 | N/A |
| 9 | chr1:g.202719900G>A | c.1816C>T | p.Arg606Ter | De novo | Nonsense | 36 | 2 |
| 10 | chr1:g.202702711G>A | c.3835C>T | p.Arg1279Ter | Mother negative, father not tested | Nonsense | 39 | 1 |
| 11 | chr1:g.202702711G>A | c.3835C>T | p.Arg1279Ter | De novo | Nonsense | 39 | 1 |
| 12 | chr1:g.202727602G>A | c.1222C>T | p.Gln408Ter | De novo | Nonsense | 38 | N/A |
| 13 | chr1:202731824:CACA:CA Deletion (2bp)a | c.1026+2_1026+3del | N/A | De novo | Splicing | N/A | N/A |
| 14 | chr1:g.202725483T>C | c.1464+3A>G | N/A | De novo | Splicing | 23.2 | N/A |
| 15 | chr1:g.202711876TTGTC>T | c.2485_2488del | p.Asp829MetfsTer9 | Inherited from affected father | Frameshift | N/A | N/A |
| 16 | chr1:g.202733231_202733240del | c.853_862del | p.Thr285GlufsTer5 | De novo | Frameshift | N/A | N/A |
| 17 | chr1:g.202704625insA | c.3463_3464insT | p.Ser1155MetfsTer9 | De novo | Frameshift | N/A | N/A |
| 18 | chr1:g.202742411AACTdel | c.408_411del | p.Val137GlnfsTer54 | Inherited from affected mother | Frameshift | N/A | N/A |
| 19 | chr1:g.202320001_203070000del | N/A | N/A | De novo | Whole gene deletion | N/A | N/A |
| 20 | chr1:g.202709936G>A | c.3058C>T | p.Arg1020Ter | De novo | Nonsense | 44 | 2 |
| chr1:g.202702855A>G | c.3691T>C | p.Cys1231Arg | Inherited | Missense | 29.3 | N/A | |
| 21 | chr1:g.202777430C>A | c.4G>T | p.Glu2Ter | Inherited from unaffected parents | Nonsense | 37 | N/A |
| chr1:g.202729568C>T | c.1160G>A | p.Trp387Ter | Nonsense | 41 | N/A |
| (A) Clinical Findings among Individuals with Dominant KDM5B Variants in this Study vs. in the Literature | ||
| Clinical Finding | This Study (n = 19) | Literature (n = 6) |
| ID | 42% | 50% |
| DD | 68% | 83% |
| Either ID or DD | 79% | 83% |
| Autistic behaviors | 53% | 33% |
| Behavioral abnormalities | 37% | 33% |
| ADHD | 16% | 0% |
| Other neurological findings | 26% | 83% |
| Renal anomalies | 21% | 17% |
| Skin anomalies | 16% | 0% |
| Finger anomalies | 26% | 17% |
| Facial dysmorphisms | 68% | 50% |
| Sleep disorder | 47% | 0% |
| Joint hypermobility | 21% | 0% |
| Ophthalmologic anomalies | 16% | 17% |
| Cardiac anomalies | 11% | 0% |
| (B) Clinical Findings among Individuals with Bi-Allelic KDM5B Variants in This Study vs. in the Literature | ||
| Clinical Finding | This Study (n = 2) | Literature (n = 8) |
| ID | 100% | 75% |
| DD | 50% | 100% |
| Either ID or DD | 100% | 100% |
| Autistic behaviors | 0% | 13% |
| Behavioral abnormalities | 100% | 0% |
| ADHD | 0% | 13% |
| Other neurological findings | 50% | 50% |
| Renal anomalies | 0% | 0% |
| Skin anomalies | 0% | 0% |
| Finger anomalies | 50% | 38% |
| Facial dysmorphisms | 100% | 88% |
| Sleep disorder | 0% | 13% |
| Joint hypermobility | 50% | 13% |
| Ophthalmologic anomalies | 50% | 38% |
| Cardiac anomalies | 0% | 25% |
| (C) Clinical Findings among Individuals with Missense vs. Disruptive Dominant KDM5B Variants in This Study | ||
| Clinical Finding | Individuals with Missense Variants (n = 8) | Individuals with Disruptive Variants (n = 11) |
| ID | 50% | 36% |
| DD | 38% | 91% |
| Either ID or DD | 63% | 91% |
| Autistic behaviors | 38% | 64% |
| Behavioral abnormalities | 38% | 36% |
| ADHD | 13% | 18% |
| Other neurological findings | 25% | 27% |
| Renal anomalies | 38% | 9% |
| Skin anomalies | 38% | 0% |
| Finger anomalies | 25% | 27% |
| Facial dysmorphisms | 63% | 73% |
| Sleep disorder | 38% | 55% |
| Joint hypermobility | 25% | 18% |
| Ophthalmologic anomalies | 13% | 18% |
| Cardiac anomalies | 13% | 9% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borroto, M.C.; Michaud, C.; Hudon, C.; Agrawal, P.B.; Agre, K.; Applegate, C.D.; Beggs, A.H.; Bjornsson, H.T.; Callewaert, B.; Chen, M.-J.; et al. A Genotype/Phenotype Study of KDM5B-Associated Disorders Suggests a Pathogenic Effect of Dominantly Inherited Missense Variants. Genes 2024, 15, 1033. https://doi.org/10.3390/genes15081033
Borroto MC, Michaud C, Hudon C, Agrawal PB, Agre K, Applegate CD, Beggs AH, Bjornsson HT, Callewaert B, Chen M-J, et al. A Genotype/Phenotype Study of KDM5B-Associated Disorders Suggests a Pathogenic Effect of Dominantly Inherited Missense Variants. Genes. 2024; 15(8):1033. https://doi.org/10.3390/genes15081033
Chicago/Turabian StyleBorroto, Maria Carla, Coralie Michaud, Chloé Hudon, Pankaj B. Agrawal, Katherine Agre, Carolyn D. Applegate, Alan H. Beggs, Hans T. Bjornsson, Bert Callewaert, Mei-Jan Chen, and et al. 2024. "A Genotype/Phenotype Study of KDM5B-Associated Disorders Suggests a Pathogenic Effect of Dominantly Inherited Missense Variants" Genes 15, no. 8: 1033. https://doi.org/10.3390/genes15081033