
DNA Methylation of Postnatal Liver Development in Pigs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Experiment and Sample Preparation
2.3. RNA-Seq
2.4. Library Preparation and Bisulfite Sequencing
2.5. Sequencing Read Quality Control and Alignment
2.6. Detection and Analysis of Methylation Sites
2.7. Correlation Analysis and PCA
2.8. DMR Analysis
2.9. Functional Enrichment Analysis for Genes
3. Results
3.1. Data Quality of BS-Seq Data
3.2. Methylation Distribution Characteristics
3.3. Characterization of Porcine Liver Transcriptome
3.4. Methylation Profile of Porcine Liver
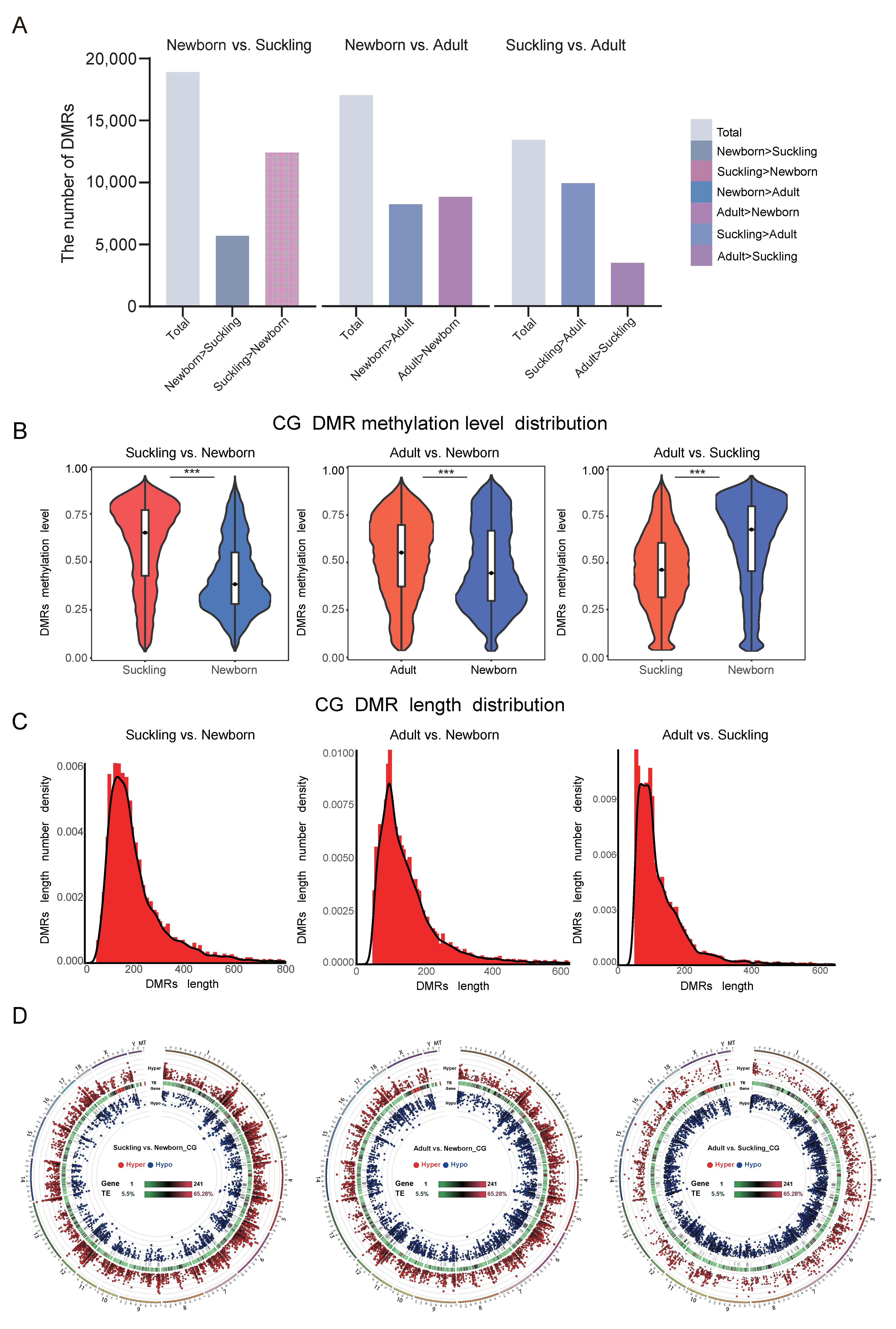
3.4.1. Distribution Characteristics of DMRs
3.4.2. The DMRs of the Promoter
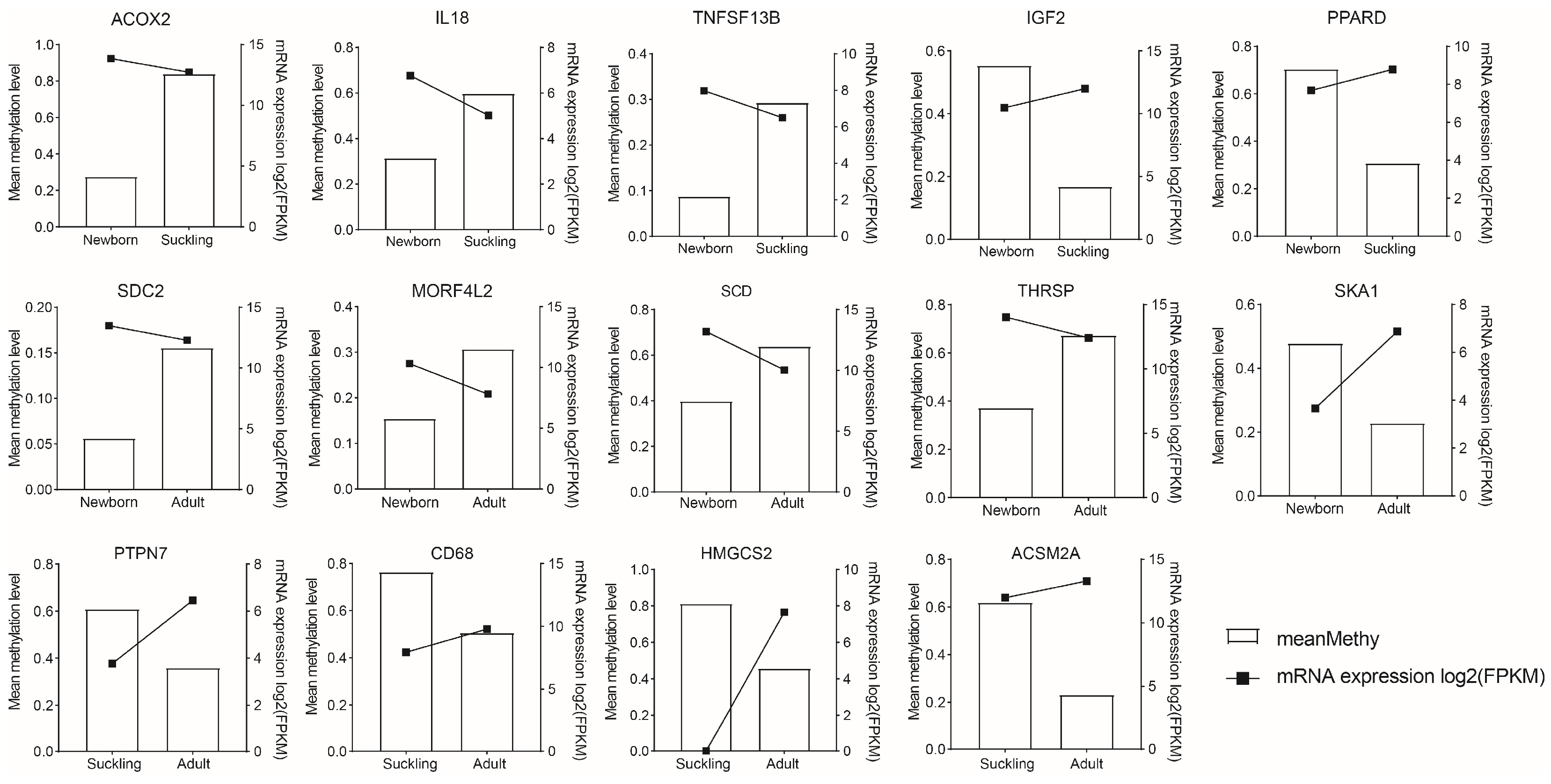
3.4.3. Genes with Differential Expression in the Promoter DMR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef]
- Bestor, T.H.; Edwards, J.R.; Boulard, M. Notes on the role of dynamic DNA methylation in mammalian development. Proc. Natl. Acad. Sci. USA 2015, 112, 6796–6799. [Google Scholar] [CrossRef]
- Curradi, M.; Izzo, A.; Badaracco, G.; Landsberger, N. Molecular mechanisms of gene silencing mediated by DNA methylation. Mol. Cell. Biol. 2002, 22, 3157–3173. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nature reviews. Genetics 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef]
- Liang, P.; Song, F.; Ghosh, S.; Morien, E.; Qin, M.; Mahmood, S.; Fujiwara, K.; Igarashi, J.; Nagase, H.; Held, W.A. Ge-nome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC Genom. 2011, 12, 231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, F.; Feng, X.; Yang, H.; Zhu, A.; Pang, J.; Han, L.; Zhang, T.; Yao, X.; Wang, F. Genome-wide analysis of DNA Methylation profiles on sheep ovaries associated with prolificacy using whole-genome Bisulfite sequencing. BMC Genom. 2017, 18, 759. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, H.; Liu, R.; He, M.; Che, T.; Jin, L.; Deng, L.; Tian, S.; Li, Y.; Lu, H.; et al. mRNA N6-methyladenosine methylation of postnatal liver development in pig. PLoS ONE 2017, 12, e0173421. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J.; Duan, J.; Gao, X.; Zhu, W.; Lu, X.; Yang, L.; Zhang, J.; Li, G.; Ci, W.; et al. Programming and inheritance of parental DNA methylomes in mammals. Cell 2014, 157, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Habibi, E.; Brinkman, A.B.; Arand, J.; Kroeze, L.I.; Kerstens, H.H.; Matarese, F.; Lepikhov, K.; Gut, M.; Brun-Heath, I.; Hubner, N.C.; et al. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell 2013, 13, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef]
- Park, Y.; Wu, H. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 2016, 32, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xu, T.; Feng, H.; Chen, L.; Li, B.; Yao, B.; Qin, Z.; Jin, P.; Conneely, K.N. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 2015, 43, e141. [Google Scholar] [CrossRef]
- Feng, H.; Conneely, K.N.; Wu, H. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014, 42, e69. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Jiang, Z.; Xia, Y.; Lou, P.; Chen, L.; Wang, H.; Bai, L.; Xie, Y.; Liu, Y.; Li, W.; et al. Genome-wide DNA methylation changes in skeletal muscle between young and middle-aged pigs. BMC Genom. 2014, 15, 653. [Google Scholar] [CrossRef]
- Jin, L.; Mao, K.; Li, J.; Huang, W.; Che, T.; Fu, Y.; Tang, Q.; Liu, P.; Song, Y.; Liu, R.; et al. Genome-wide profiling of gene expression and DNA methylation provides insight into low-altitude acclimation in Tibetan pigs. Gene 2018, 642, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.A.; Shimbo, T.; Takaku, M.; Thomas, J.W.; Auerbach, S.; Bennett, B.D.; Bucher, J.R.; Burkholder, A.B.; Day, F.; Du, Y.; et al. DNA methylation in mice is influenced by genetics as well as sex and life experience. Nat. Commun. 2019, 10, 305. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, L.; Huang, Y.; Shen, F.; Wu, H.; Yang, Z.; Hou, R.; Song, Z.; Yue, B.; Zhang, X. Epigenomic profiling indicates a role for DNA methylation in the postnatal liver and pancreas development of giant pandas. Genomics 2022, 114, 110342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N. Role of methionine on epigenetic modification of DNA methylation and gene expression in animals. Anim. Nutr. (Zhongguo Xu Mu Shou Yi Xue Hui) 2018, 4, 11–16. [Google Scholar] [CrossRef]
- Cao, J.; Wei, C.; Liu, D.; Wang, H.; Wu, M.; Xie, Z.; Capellini, T.D.; Zhang, L.; Zhao, F.; Li, L.; et al. DNA methylation Landscape of body size variation in sheep. Sci. Rep. 2015, 5, 13950. [Google Scholar] [CrossRef]
- Gutierrez-Arcelus, M.; Lappalainen, T.; Montgomery, S.B.; Buil, A.; Ongen, H.; Yurovsky, A.; Bryois, J.; Giger, T.; Romano, L.; Planchon, A.; et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife 2013, 2, e00523. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Williams, J.; Xu, B.; Putnam, D.; Thrasher, A.; Li, C.; Yang, J.; Chen, X. MethylationToActivity: A deep-learning framework that reveals promoter activity landscapes from DNA methylomes in individual tumors. Genome Biol. 2021, 22, 24. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Y.; Huang, R.; Song, W.; Wang, J.; Xiao, Z.; Dong, S.; Yang, Y.; Yang, X. Dependency of the Cancer-Specific Transcriptional Regulation Circuitry on the Promoter DNA Methylome. Cell Rep. 2019, 26, 3461–3474.e5. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. Expression of various genes is controlled by DNA methylation during mammalian development. J. Cell. Biochem. 2003, 88, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Sabag, O.; Reizel, Y. The role of DNA methylation in genome-wide gene regulation during development. Development 2022, 149, dev200118. [Google Scholar] [CrossRef] [PubMed]
- Huse, S.M.; Gruppuso, P.A.; Boekelheide, K.; Sanders, J.A. Patterns of gene expression and DNA methylation in human fetal and adult liver. BMC Genom. 2015, 16, 981. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Muroya, S.; Otomaru, K.; Oshima, K.; Oshima, I.; Ojima, K.; Gotoh, T. DNA Methylation of Genes Participating in Hepatic Metabolisms and Function in Fetal Calf Liver Is Altered by Maternal Undernutrition during Gestation. Int. J. Mol. Sci. 2023, 24, 10682. [Google Scholar] [CrossRef] [PubMed]
- Ehara, T.; Kamei, Y.; Yuan, X.; Takahashi, M.; Kanai, S.; Tamura, E.; Tsujimoto, K.; Tamiya, T.; Nakagawa, Y.; Shimano, H.; et al. Ligand-activated PPARα-dependent DNA demethylation regulates the fatty acid β-oxidation genes in the postnatal liver. Diabetes 2015, 64, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hao, S.; Shi, Q.; Deng, Q.; Jiang, Y.; Guo, P.; Yuan, Y.; Shi, X.; Shangguan, S.; Zheng, H.; et al. Transcriptomic Profile of the Mouse Postnatal Liver Development by Single-Nucleus RNA Sequencing. Front. Cell Dev. Biol. 2022, 10, 833392. [Google Scholar] [CrossRef]
- Drackley, J.K. ADSA Foundation Scholar Award. Biology of dairy cows during the transition period: The final frontier? J. Dairy Sci. 1999, 82, 2259–2273. [Google Scholar] [CrossRef]
- van Milgen, J.; Noblet, J. Partitioning of energy intake to heat, protein, and fat in growing pigs. J. Anim. Sci. 2003, 81, E86–E93. [Google Scholar] [CrossRef]
- Rao, L.; Cai, L.; Huang, L. Single-cell dynamics of liver development in postnatal pigs. Sci. Bull. 2023, 68, 2583–2597. [Google Scholar] [CrossRef] [PubMed]
- Schiöth, H.B.; Boström, A.; Murphy, S.K.; Erhart, W.; Hampe, J.; Moylan, C.; Mwinyi, J. A targeted analysis reveals relevant shifts in the methylation and transcription of genes responsible for bile acid homeostasis and drug metabolism in non-alcoholic fatty liver disease. BMC Genom. 2016, 17, 462. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Jiang, Z.; Zhou, J. The role of lipin-1 in the pathogenesis of alcoholic fatty liver. Alcohol Alcohol. 2015, 50, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Gutierrez, M.J.; Nino, G.; Hong, X.; Wang, X. Epigenetic Dynamics of the Infant Immune System Reveals a Tumor Necrosis Factor Superfamily Signature in Early Human Life. Epigenomes 2020, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Duodu, P.; Sosa, G.; Canar, J.; Chhugani, O.; Gamero, A.M. Exposing the Two Contrasting Faces of STAT2 in Inflammation. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2022, 42, 467–481. [Google Scholar] [CrossRef]
- Nair, S.; Bist, P.; Dikshit, N.; Krishnan, M.N. Global functional profiling of human ubiquitome identifies E3 ubiquitin ligase DCST1 as a novel negative regulator of Type-I interferon signaling. Sci. Rep. 2016, 6, 36179. [Google Scholar] [CrossRef]
- Yamanishi, K.; Hata, M.; Gamachi, N.; Watanabe, Y.; Yamanishi, C.; Okamura, H.; Matsunaga, H. Molecular Mechanisms of IL18 in Disease. Int. J. Mol. Sci. 2023, 24, 17170. [Google Scholar] [CrossRef]
- Wang, H.; Liu, B.; Wei, J. Beta2-microglobulin(B2M) in cancer immunotherapies: Biological function, resistance and remedy. Cancer Lett. 2021, 517, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Inamdar, V.V.; Reddy, H.; Dangelmaier, C.; Kostyak, J.C.; Kunapuli, S.P. The protein tyrosine phosphatase PTPN7 is a negative regulator of ERK activation and thromboxane generation in platelets. J. Biol. Chem. 2019, 294, 12547–12554. [Google Scholar] [CrossRef] [PubMed]
- Tousson, E.; Beltagy, D.M.; Gazia, M.A.; Al-Behbehani, B. Expressions of P53 and CD68 in mouse liver with Schistosoma mansoni infection and the protective role of silymarin. Toxicol. Ind. Health 2013, 29, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Webb, L.M.; Datta, P.; Bell, S.E.; Kitamura, D.; Turner, M.; Butcher, G.W. GIMAP1 Is Essential for the Survival of Naive and Activated B Cells In Vivo. J. Immunol. 2016, 196, 207–216. [Google Scholar] [CrossRef]
- Freitas-Lopes, M.A.; Mafra, K.; David, B.A.; Carvalho-Gontijo, R.; Menezes, G.B. Differential location and distribution of hepatic immune cells. Cells 2017, 6, 48. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Jin, H.; Tong, X.; Yu, H.; Li, X.; Zeng, B. DNA Methylation of Postnatal Liver Development in Pigs. Genes 2024, 15, 1067. https://doi.org/10.3390/genes15081067
Wang Y, Jin H, Tong X, Yu H, Li X, Zeng B. DNA Methylation of Postnatal Liver Development in Pigs. Genes. 2024; 15(8):1067. https://doi.org/10.3390/genes15081067
Chicago/Turabian StyleWang, Yuhao, Hongling Jin, Xingyan Tong, Huan Yu, Xuewei Li, and Bo Zeng. 2024. "DNA Methylation of Postnatal Liver Development in Pigs" Genes 15, no. 8: 1067. https://doi.org/10.3390/genes15081067
APA StyleWang, Y., Jin, H., Tong, X., Yu, H., Li, X., & Zeng, B. (2024). DNA Methylation of Postnatal Liver Development in Pigs. Genes, 15(8), 1067. https://doi.org/10.3390/genes15081067