

Proteomic and Phosphoproteomic Analyses during Plant Regeneration Initiation in Cotton (Gossypium hirsutum L.)
 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Positive Correlation between Changes in Proteins and Their Phosphorylation
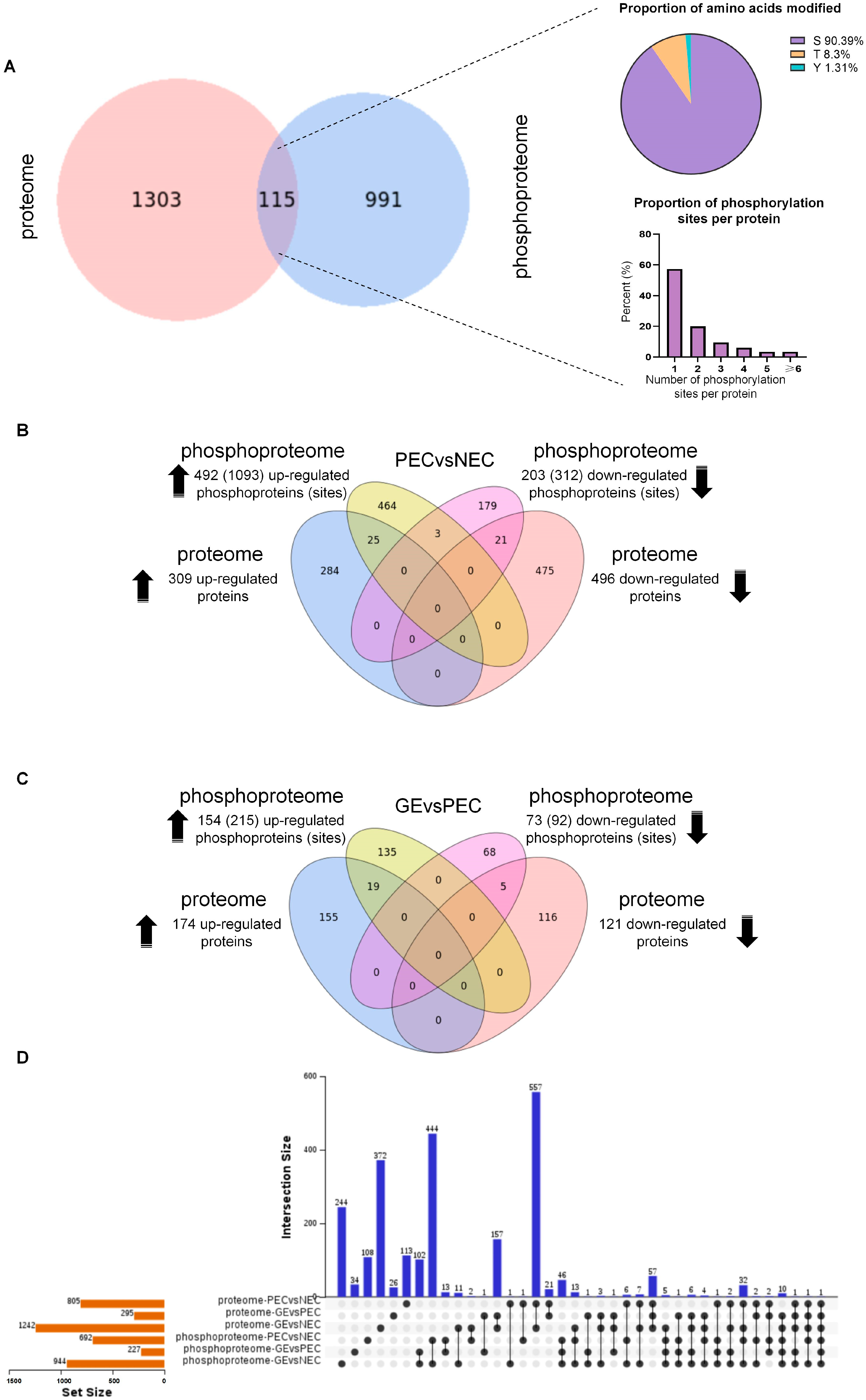
2.2. Association Analysis of Differentially Accumulated Proteins (DAPs) and Correlated Differentially Regulated Phosphoproteins (DRPPs)
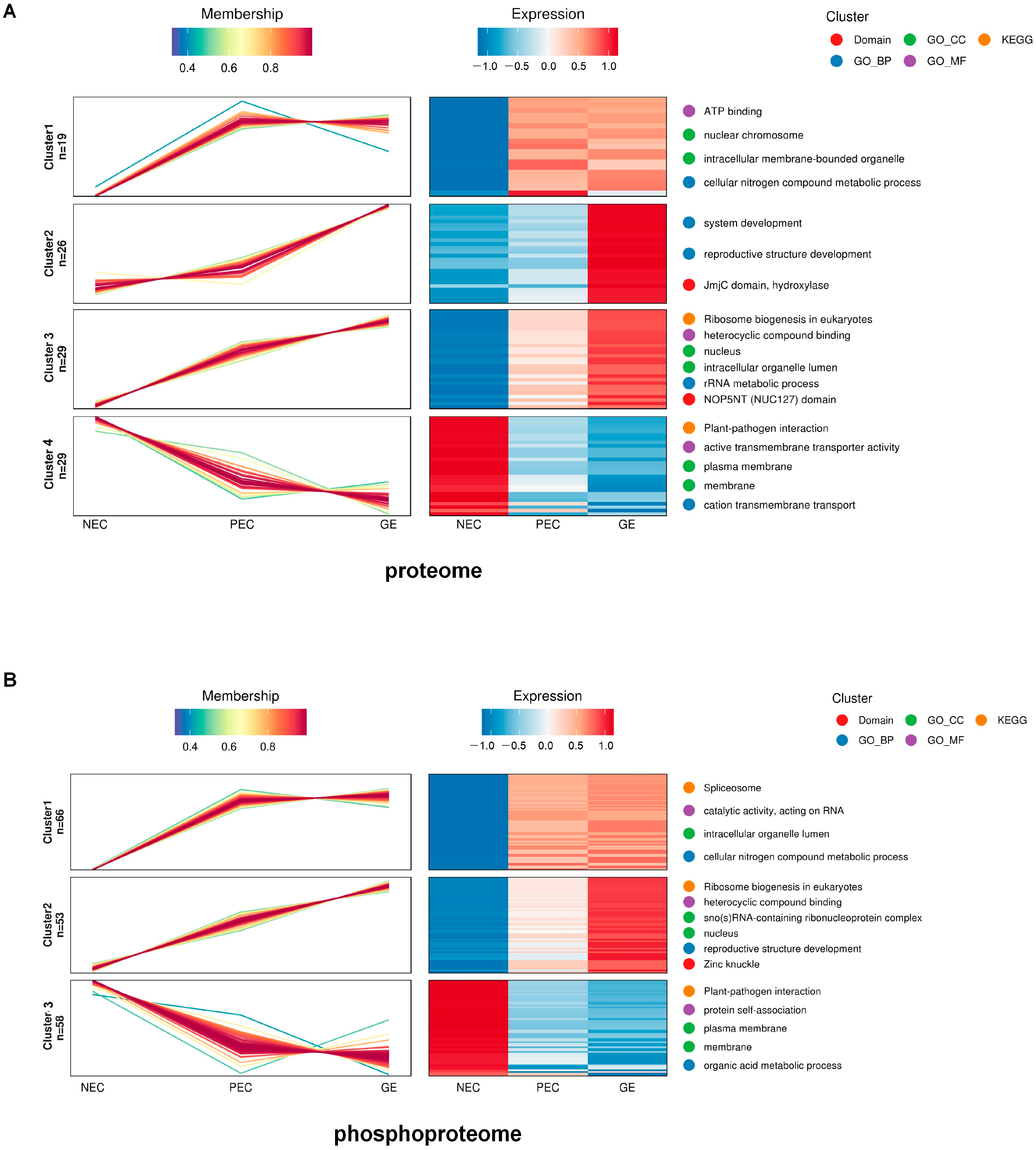
2.3. Dynamic Trajectory Analysis of Overlapping Proteins (DAPs-DRPPs) during Plant Regeneration Initiation
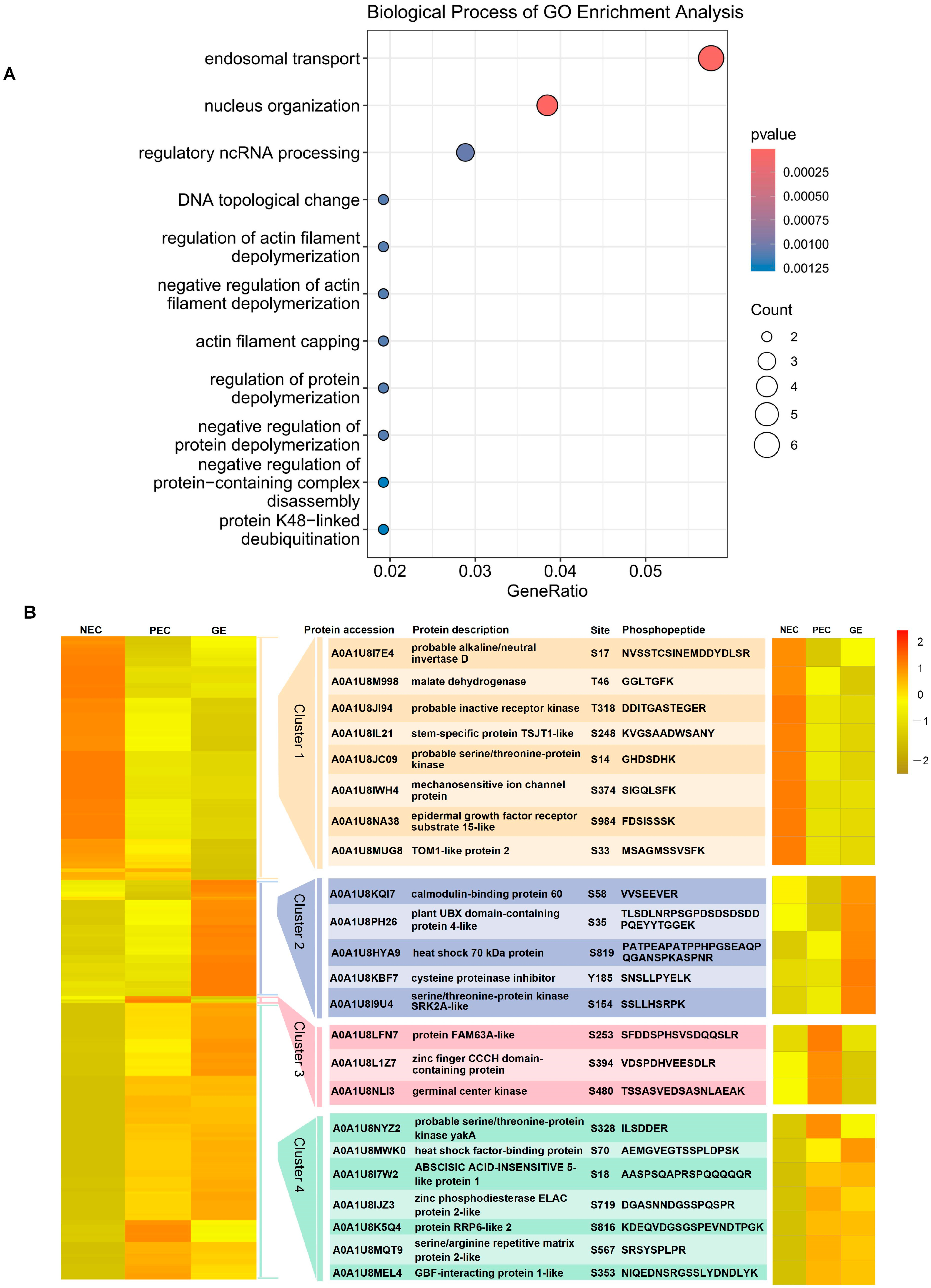
2.4. Enrichment Analysis of Proteins Susceptible to Regulation by Phosphorylation
3. Conclusions
4. Discussion
4.1. DAPs-DRPPs with Different Trajectory Patterns during Plant Regeneration Initiation
4.2. Proteins Susceptible to Regulation by Phosphorylation
4.3. Potential Critical DAPs-DRPPs in the Initiation of Plant Regeneration
5. Materials and Method
5.1. Association Analysis of the Proteome and Phosphoproteome
5.2. Analysis of DAPs and Correlated DRPPs (Differential Protein Statistics)
5.3. Hierarchical Clustering Analysis
5.4. Functional Enrichment Analysis
5.5. Enrichment-Based Clustering
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vogel, G. How does a single somatic cell become a whole plant? Science 2005, 309, 86. [Google Scholar] [CrossRef]
- Roeder, A.; Otegui, M.; Dixit, R.; Anderson, C.; Faulkner, C.; Zhang, Y.; Harrison, M.; Kirchhelle, C.; Goshima, G.; Coate, J.; et al. Fifteen compelling open questions in plant cell biology. Plant Cell 2021, 34, 72–102. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Xiang, D.; Wu, N.; Wang, Y.; Chen, Y.; Yuan, Y.; Ye, Y.; Hu, D.; Zheng, C.; Yan, Y.; et al. Control of rice ratooning ability by a nucleoredoxin that inhibits histidine kinase dimerization to attenuate cytokinin signaling in axillary buds. Mol. Plant 2023, 16, 1911–1926. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, C.; Zhang, X.; Liu, M.; Xue, X.; Lai, C.; Xu, X.; Chen, Y.; Zhang, Z.; Lai, Z.; et al. Single-cell RNA sequencing analysis of the embryogenic callus clarifies the spatiotemporal developmental trajectories of the early somatic embryo in Dimocarpus longan. Plant J. 2023, 115, 1277–1297. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xu, Z.; Wang, G.; Cong, Y.; Yu, L.; Jia, R.; Qin, Y.; Zhang, G.; Li, B.; Yuan, D.; et al. Single-cell resolution analysis reveals the preparation for reprogramming the fate of stem cell niche in cotton lateral meristem. Genome Biol. 2023, 24, 194. [Google Scholar]
- Yang, X.; Zhang, X. Regulation of somatic embryogenesis in higher plants. Crit. Rev. Plant Sci. 2010, 29, 36–57. [Google Scholar] [CrossRef]
- Zhai, N.; Xu, L. Pluripotency acquisition in the middle cell layer of callus is required for organ regeneration. Nat. Plants 2021, 7, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Fehér, A. Somatic embryogenesis-Stress-induced remodeling of plant cell fate. Biochim. Biophys. Acta 2015, 1849, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Liang, S.; Zhang, X.; Nie, Y.; Guo, X. An efficient grafting system for transgenic plant recovery in cotton (Gossypium hirsutum L.). Plant Cell Tissue Organ. Cult. 2006, 85, 181–185. [Google Scholar] [CrossRef]
- Cheng, W.; Zhu, H.; Tian, W.; Zhu, S.; Xiong, X.; Sun, Y.; Zhu, Q.; Sun, J. De novo transcriptome analysis reveals insights into dynamic homeostasis regulation of somatic embryogenesis in upland cotton (G. hirsutum L.). Plant Mol. Biol. 2016, 92, 279–292. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Yuan, D.; Jin, F.; Zhang, Y.; Xu, J. Transcript profiling reveals complex auxin signalling pathway and transcription regulation involved in dedifferentiation and redifferentiation during somatic embryogenesis in cotton. BMC Plant Biol. 2012, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Hu, L.; Yuan, D.; Xu, J.; Gao, W.; He, L.; Yang, X.; Zhang, X. Comparative transcriptome analysis between somatic embryos (SEs) and zygotic embryos in cotton: Evidence for stress response functions in SE development. Plant Biotechnol. J. 2014, 12, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Sakhanokho, H.; Rajasekaran, K. Cotton Regeneration In Vitro. In Fiber Plants. Sustainable Development and Biodiversity; Ramawat, K., Ahuja, M., Eds.; Springer: Cham, Switzerland, 2016; Volume 13. [Google Scholar]
- Li, J.; Wang, M.; Li, Y.; Zhang, Q.; Lindsey, K.; Daniell, H.; Jin, S.; Zhang, X. Multi-omics analyses reveal epigenomics basis for cotton somatic embryogenesis through successive regeneration acclimation process. Plant Biotechnol. J. 2019, 17, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, T.; Mishra, R.; Trolinder, N. Agrobacterium-mediated transformation and regeneration of cotton. J. Food Agric. Environ. 2004, 2, 179–187. [Google Scholar]
- Bouchabke-Coussa, O.; Obellianne, M.; Linderme, D.; Montes, E.; Maia-Grondard, A.; Vilaine, F.; Pannetier, C. Wuschel overexpression promotes somatic embryogenesis and induces organogenesis in cotton (Gossypium hirsutum L.) tissues cultured in vitro. Plant Cell Rep. 2013, 32, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Cheng, W.; Tian, W.; Li, Y.; Liu, F.; Xue, F.; Zhu, Q.; Sun, Y.; Sun, J. iTRAQ-based comparative proteomic analysis provides insights into somatic embryogenesis in Gossypium hirsutum L. Plant Mol. Biol. 2018, 96, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Du, Q.; Tian, C.; Wang, Y.; Jiao, Y. Stochastic gene expression drives mesophyll protoplast regeneration. Sci. Adv. 2021, 7, eabg8466. [Google Scholar] [CrossRef] [PubMed]
- Ogura, N.; Sasagawa, Y.; Ito, T.; Tameshige, T.; Kawai, S.; Sano, M.; Doll, Y.; Iwase, A.; Kawamura, A.; Suzuki, T.; et al. WUSCHEL-RELATED HOMEOBOX 13 suppresses de novo shoot regeneration via cell fate control of pluripotent callus. Sci. Adv. 2023, 9, eadg6983. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bie, X.; Lin, X.; Li, M.; Wang, H.; Zhang, X.; Yang, Y.; Zhang, C.; Zhang, X.; Xiao, J. Uncovering the transcriptional regulatory network involved in boosting wheat regeneration and transformation. Nat. Plants 2023, 9, 908–925. [Google Scholar] [CrossRef]
- Xu, C.; Chang, P.; Guo, S.; Yang, X.; Liu, X.; Sui, B.; Yu, D.; Xin, W.; Hu, Y. Transcriptional activation by WRKY23 and derepression by removal of bHLH041 coordinately establish callus pluripotency in Arabidopsis regeneration. Plant Cell 2023, 36, 158–173. [Google Scholar] [CrossRef]
- Ge, X.; Zhang, C.; Wang, Q.; Yang, Z.; Wang, Y.; Zhang, X.; Wu, Z.; Hou, Y.; Wu, J.; Li, F. iTRAQ protein profile differential analysis between somatic globular and cotyledonary embryos reveals stress, hormone, and respiration involved in increasing plantlet regeneration of Gossypium hirsutum L. J. Proteome Res. 2015, 14, 268–278. [Google Scholar] [CrossRef]
- Juturu, V.; Mekala, G.; Kirti, P. Current status of tissue culture and genetic transformation research in cotton (Gossypium spp.). Plant Cell Tissue Organ. Cult. 2015, 120, 813–839. [Google Scholar] [CrossRef]
- Min, L.; Hu, Q.; Li, Y.; Xu, J.; Ma, Y.; Zhu, L.; Yang, X.; Zhang, X. LEAFY COTYLEDON1-CASEIN KINASE I-TCP15-PHYTOCHROME INTERACTING FACTOR4 Network Regulates Somatic Embryogenesis by Regulating Auxin Homeostasis. Plant Physiol. 2015, 169, 2805–2821. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Shang, G.; Wang, F.; Gao, J.; Wan, M.; Xu, Z.; Wang, J. Dynamic chromatin state profiling reveals regulatory roles of auxin and cytokinin in shoot regeneration. Dev. Cell 2022, 57, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, L.; Yuan, D.; Lindsey, K.; Zhang, X. Small RNA and degradome sequencing reveal complex miRNA regulation during cotton somatic embryogenesis. J. Exp. Bot. 2013, 64, 1521–1536. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Fan, Y.; Guo, H.; Wu, J.; Yu, X.; Wei, J.; Lian, X.; Zhang, L.; Gou, Z.; Fan, Y.; et al. Somatic embryogenesis critical initiation stage-specific mCHH hypomethylation reveals epigenetic basis underlying embryogenic redifferentiation in cotton. Plant Biotechnol. J. 2020, 18, 1648–1650. [Google Scholar] [CrossRef]
- Zhang, S.; Yu, R.; Yu, D.; Chang, P.; Guo, S.; Yang, X.; Liu, X.; Xu, C.; Hu, Y. The calcium signaling module CaM-IQM destabilizes IAA-ARF interaction to regulate callus and lateral root formation. Proc. Natl. Acad. Sci. USA 2022, 119, e2202669119. [Google Scholar] [CrossRef]
- Yu, Y.; Yu, H.; Peng, J.; Yao, W.; Wang, Y.; Zhang, F.; Wang, S.; Zhao, Y.; Zhao, X.; Zhang, X.; et al. Enhancing wheat regeneration and genetic transformation through overexpression of TaLAX1. Plant Commun. 2023, 5, 100738. [Google Scholar] [CrossRef]
- Avila, C.; Suárez, M.; Gómez-Maldonado, J.; Cánovas, F. Spatial and temporal expression of two cytosolic glutamine synthetase genes in Scots pine: Functional implications on nitrogen metabolism during early stages of conifer development. Plant J. 2001, 25, 93–102. [Google Scholar] [CrossRef]
- Rodríguez, M.; Suárez, M.; Heredia, R.; Avila, C.; Breton, D.; Trontin, J.; Filonova, L.; Bozhkov, P.; von Arnold, S.; Harvengt, L.; et al. Expression patterns of two glutamine synthetase genes in zygotic and somatic pine embryos support specific roles in nitrogen metabolism during embryogenesis. New Phytol. 2006, 169, 35–44. [Google Scholar] [CrossRef]
- Mao, X.; Zhang, H.; Tian, S.; Chang, X.; Jing, R. TaSnRK2.4, an SNF1-type serine/threonine protein kinase of wheat (Triticum aestivum L.), confers enhanced multistress tolerance in Arabidopsis. J. Exp. Bot. 2010, 61, 683–696. [Google Scholar] [CrossRef]
- Korbei, B.; Moulinier-Anzola, J.; De-Araujo, L.; Lucyshyn, D.; Retzer, K.; Khan, M.; Luschnig, C. Arabidopsis TOL proteins act as gatekeepers for vacuolar sorting of PIN2 plasma membrane protein. Curr. Biol. 2013, 23, 2500–2505. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; van Kleeff, P.; Oecking, C.; Li, K.; Erban, A.; Kopka, J.; Hincha, D.; de Boer, A. Light modulated activity of root alkaline/neutral invertase involves the interaction with 14-3-3 proteins. Plant J. 2014, 80, 785–796. [Google Scholar] [CrossRef]
- Zhang, H.; Tang, K.; Qian, W.; Duan, C.; Wang, B.; Zhang, H.; Wang, P.; Zhu, X.; Lang, Z.; Yang, Y.; et al. An Rrp6-like protein positively regulates noncoding RNA levels and DNA methylation in Arabidopsis. Mol. Cell 2014, 54, 418–430. [Google Scholar] [CrossRef]
- Tang, D.; Quan, C.; Lin, Y.; Wei, K.; Qin, S.; Liang, Y.; Wei, F.; Miao, J. Physio-Morphological, Biochemical and Transcriptomic Analyses Provide Insights into Drought Stress Responses in Mesona chinensis Benth. Front. Plant Sci. 2022, 13, 809723. [Google Scholar] [CrossRef]
- Maris, C.; Dominguez, C.; Allain, F. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005, 272, 2118–2131. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Fiers, M.; Dekkers, B.; Maas, L.; van Esse, G.; Angenent, G.; Zhao, Y.; Boutilier, K. ABA signalling promotes cell totipotency in the shoot apex of germinating embryos. J. Exp. Bot. 2021, 72, 6418–6436. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Yan, Y.; Wang, Y.; Liu, S.; Guo, W.; Yang, L.; Shen, H. Proper doses of brassinolide enhance somatic embryogenesis in different competent Korean pine cell lines during embryogenic callus differentiation. Front. Plant Sci. 2024, 15, 1330103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, Y.; Cheng, Y.; Huang, J.; Lian, J.; Yang, L.; He, R.; Lei, M.; Liu, Y.; Yuan, C.; et al. Genome-wide analysis and functional annotation of chromatin-enriched non coding RNAs in rice during somatic cell regeneration. Genome Biol. 2022, 23, 28. [Google Scholar] [CrossRef]
- Gaudet, P.; Livstone, M.; Lewis, S.; Thomas, P. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Abdul, R.; Kristariyanto, Y.; Choi, S.; Nkosi, P.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 7, 146–155. [Google Scholar]
- Yan, Z.; Jia, J.; Yan, X.; Shi, H.; Han, Y. Arabidopsis KHZ1 and KHZ2, two novel non-tandem CCCH zinc-finger and K-homolog domain proteins, have redundant roles in the regulation of flowering and senescence. Plant Mol. Biol. 2017, 95, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Pi, B.; He, X.; Ruan, Y.; Jang, J.; Huang, Y. Genome-wide analysis and stress-responsive expression of CCCH zinc finger family genes in Brassica rapa. BMC Plant Biol. 2018, 18, 373. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Shi, Z.; Jiao, S.; Chen, C.; Wang, W.; Greene, M.; Zhou, Z. Germinal center kinases in immune regulation. Cell Mol. Immunol. 2012, 9, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Kamada, H. Initial identification of a phosphoprotein that appears to be involved in the induction of somatic embryogenesis in carrot. Plant Cell Rep. 2000, 19, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Burrieza, H.; López-Fernández, M.; Chiquieri, T.; Silveira, V.; Maldonado, S. Accumulation pattern of dehydrins during sugarcane (var. SP80.3280) somatic embryogenesis. Plant Cell Rep. 2012, 31, 2139–2149. [Google Scholar] [CrossRef]
- Guo, H.; Guo, H.; Zhang, L.; Fan, Y.; Fan, Y.; Tang, Z.; Zeng, F. Dynamic TMT-Based Quantitative Proteomics Analysis of Critical Initiation Process of Totipotency during Cotton Somatic Embryogenesis Transdifferentiation. Int. J. Mol. Sci. 2019, 20, 1691. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014; p. 1. [Google Scholar]
- Benesty, J.; Chen, J.; Huang, Y.; Cohen, I. Pearson Correlation Coefficient. In Noise Reduction in Speech Processing; Springer Topics in Signal Processing; Springer: Berlin/Heidelberg, Germany, 2009; Volume 2. [Google Scholar]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Wang, J.; Huo, X.; Cui, X.; Zhang, L.; Qi, X.; Wu, X.; Liu, J.; Wang, A.; Liu, J.; et al. Proteomic and Phosphoproteomic Analyses during Plant Regeneration Initiation in Cotton (Gossypium hirsutum L.). Genes 2024, 15, 1079. https://doi.org/10.3390/genes15081079
Guo H, Wang J, Huo X, Cui X, Zhang L, Qi X, Wu X, Liu J, Wang A, Liu J, et al. Proteomic and Phosphoproteomic Analyses during Plant Regeneration Initiation in Cotton (Gossypium hirsutum L.). Genes. 2024; 15(8):1079. https://doi.org/10.3390/genes15081079
Chicago/Turabian StyleGuo, Haixia, Jin Wang, Xuehui Huo, Xiwang Cui, Li Zhang, Xiushan Qi, Xiaoying Wu, Junchen Liu, Aijuan Wang, Jialin Liu, and et al. 2024. "Proteomic and Phosphoproteomic Analyses during Plant Regeneration Initiation in Cotton (Gossypium hirsutum L.)" Genes 15, no. 8: 1079. https://doi.org/10.3390/genes15081079
APA StyleGuo, H., Wang, J., Huo, X., Cui, X., Zhang, L., Qi, X., Wu, X., Liu, J., Wang, A., Liu, J., Chen, X., Zeng, F., & Guo, H. (2024). Proteomic and Phosphoproteomic Analyses during Plant Regeneration Initiation in Cotton (Gossypium hirsutum L.). Genes, 15(8), 1079. https://doi.org/10.3390/genes15081079