
Meta-Genomic Analysis of Different Bacteria and Their Genomes Found in Raw Buffalo Milk Obtained in Various Farms Using Different Milking Methods
 ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. Metagenomic Sequencing
2.4. Taxonomic Assignment, Functional Annotation and Gene Function Analysis
2.5. Statistical Analysis
3. Results and Discussion
3.1. Effect of Different Milking Methods on the Basal Nutrient Content, SCC and Number of Bacterial Cells of Raw Buffalo Milk
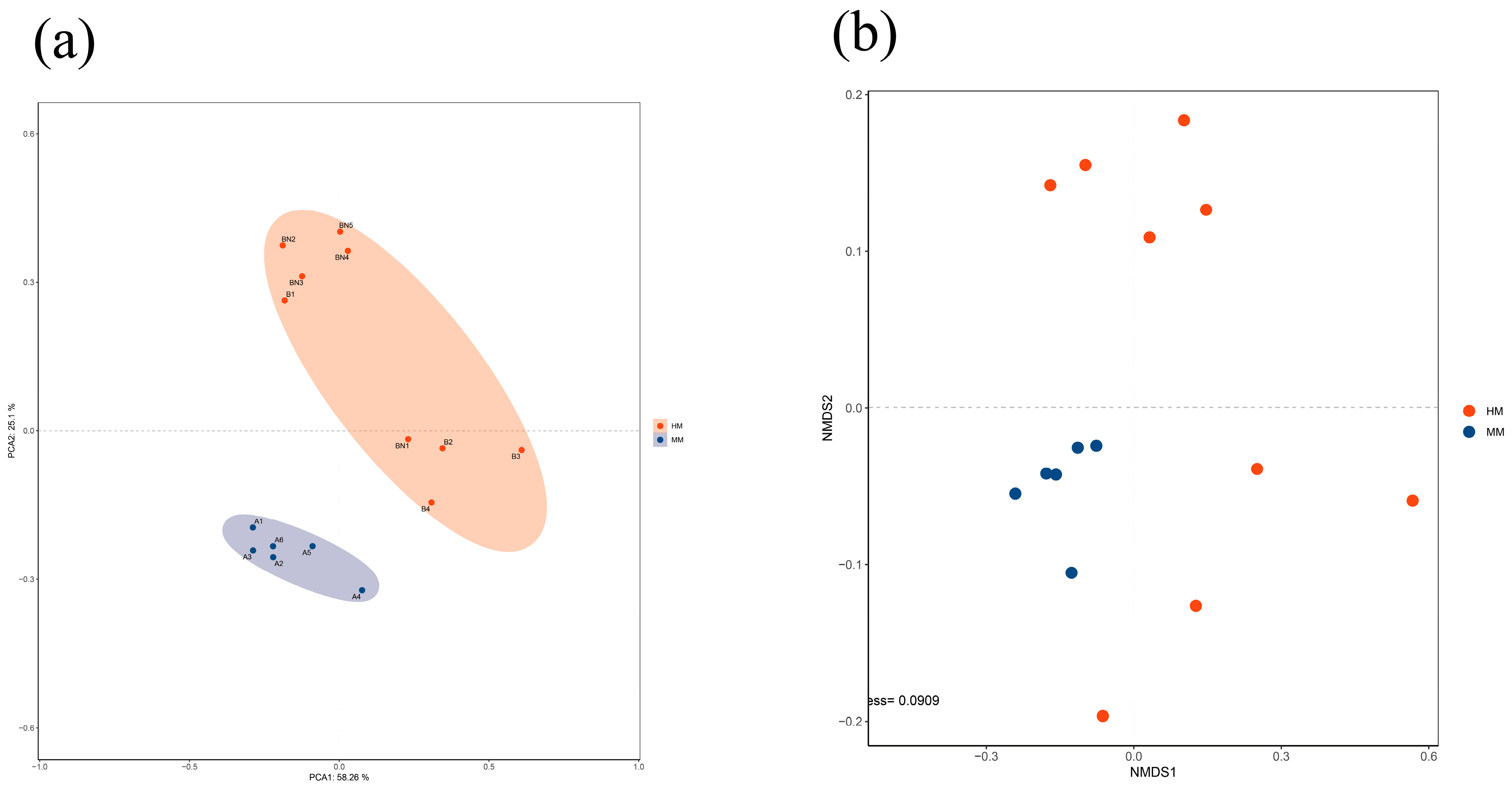
3.2. Milking Methods Influence the Raw Milk Microbiota
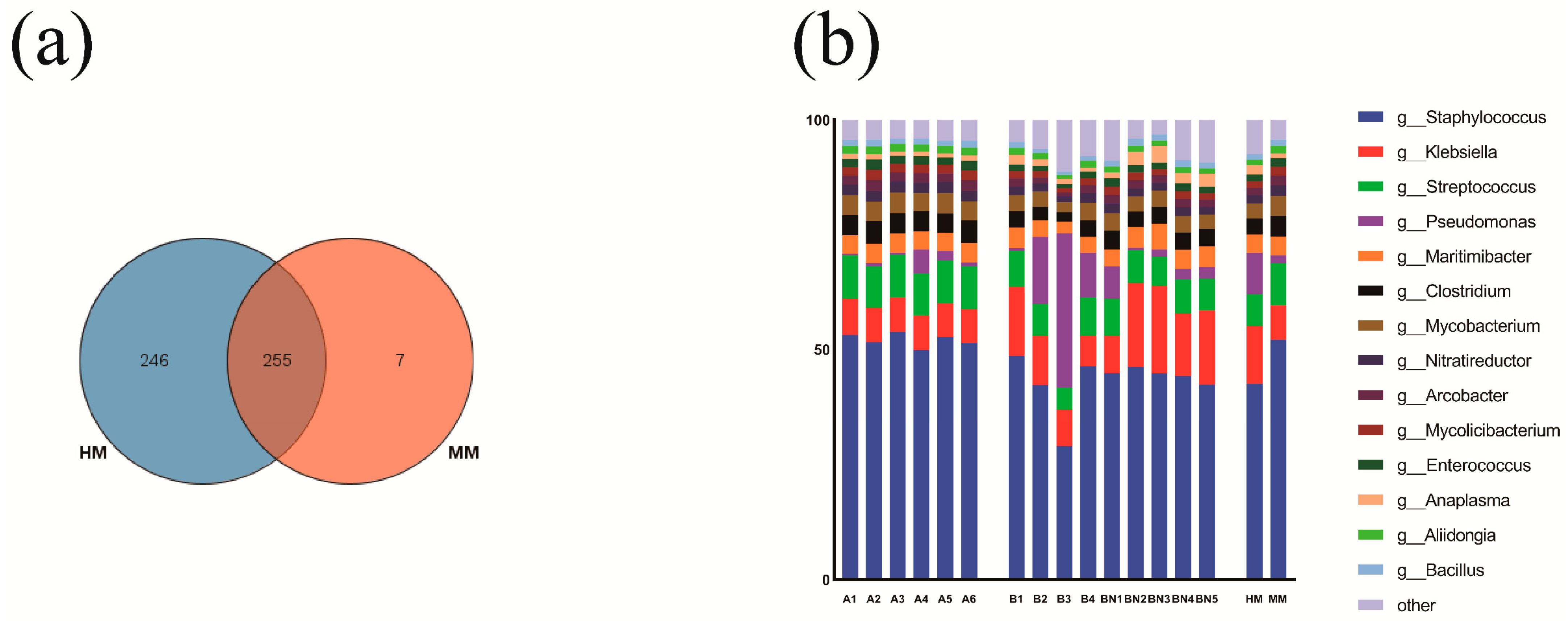
3.3. Structure and Composition of the Milk Microbiome of Buffaloes with Different Milking Methods
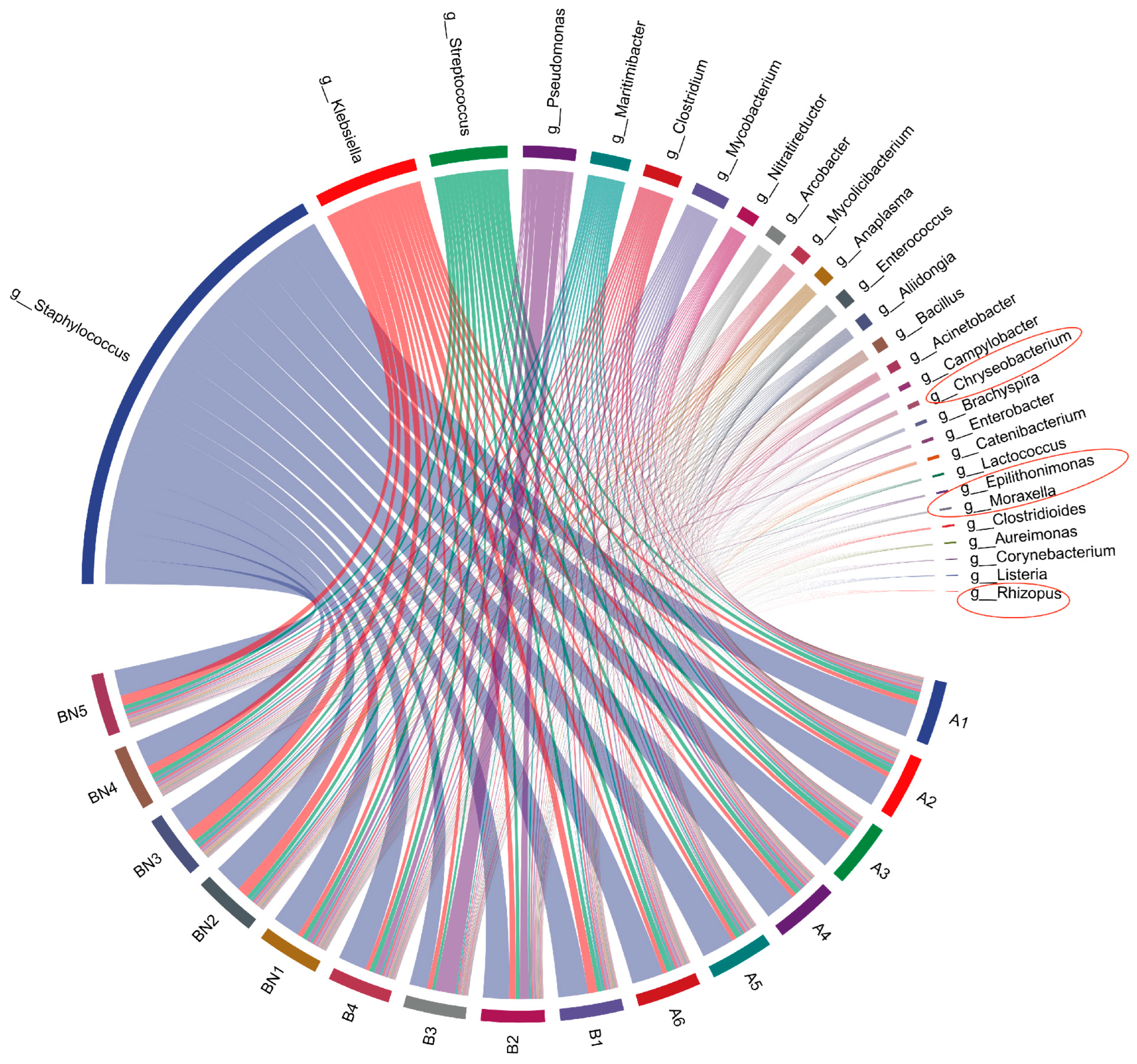
3.4. Differences in the Relative Abundance Composition of Core Microbiota and Bacteria Potentially Associated with Mastitis at the Genus Level between Milking Methods
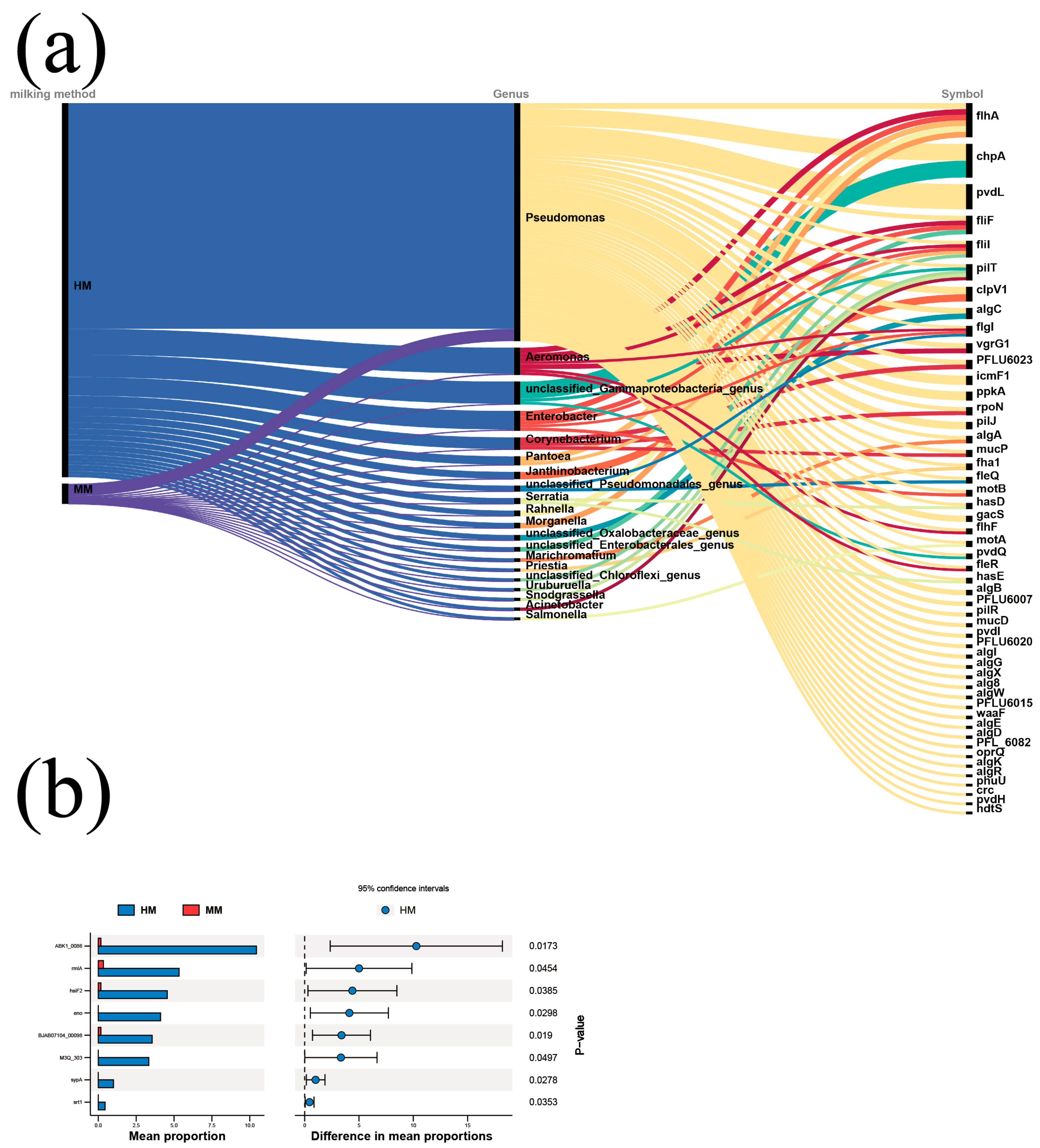
3.5. Effect of Milking Methods on Antibiotic Resistance Genes
3.6. Effect of Milking Methods on the Level of Virulence Factors of Pathogenic Bacteria
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nanda, A.S.; Nakao, T. Role of buffalo in the socioeconomic development of rural Asia: Current status and future prospectus. Anim. Sci. J. 2003, 74, 443–455. [Google Scholar] [CrossRef]
- Nie, P.; Pan, B.; Ahmad, M.J.; Zhang, X.; Chen, C.; Yao, Z.; Lv, H.; Wei, K.; Yang, L. Summer buffalo milk produced in China: A desirable diet enriched in polyunsaturated fatty acids and amino acids. Foods 2022, 11, 3475. [Google Scholar] [CrossRef]
- Quigley, L.; O’Sullivan, O.; Stanton, C.; Beresford, T.P.; Ross, R.P.; Fitzgerald, G.F.; Cotter, P.D. The complex microbiota of raw milk. FEMS Microbiol. Rev. 2013, 37, 664–698. [Google Scholar] [CrossRef] [PubMed]
- Addis, M.F.; Tanca, A.; Uzzau, S.; Oikonomou, G.; Bicalho, R.C.; Moroni, P. The bovine milk microbiota: Insights and perspectives from -omics studies. Mol. BioSyst. 2016, 12, 2359–2372. [Google Scholar] [CrossRef]
- Machado, S.G.; Baglinière, F.; Marchand, S.; Van Coillie, E.; Vanetti, M.C.D.; De Block, J.; Heyndrickx, M. The biodiversity of the microbiota producing heat-resistant enzymes responsible for spoilage in processed bovine milk and dairy products. Front. Microbiol. 2017, 8, 302. [Google Scholar] [CrossRef]
- Elmoslemany, A.M.; Keefe, G.; Dohoo, I.; Wichtel, J.; Stryhn, H.; Dingwell, R. The association between bulk tank milk analysis for raw milk quality and on-farm management practices. Prev. Vet. Med. 2010, 95, 32–40. [Google Scholar] [CrossRef]
- Abebe, R.; Hatiya, H.; Abera, M.; Megersa, B.; Asmare, K. Bovine mastitis: Prevalence, risk factors and isolation of Staphylococcus aureus in dairy herds at Hawassa milk shed, South Ethiopia. BMC Vet. Res. 2016, 12, 270. [Google Scholar] [CrossRef]
- Bhakat, C.; Mohammad, A.; Mandal, D.; Mandal, A.; Rai, S.; Chatterjee, A.; Ghosh, M.; Dutta, T. Readily usable strategies to control mastitis for production augmentation in dairy cattle: A review. Vet. World 2020, 13, 2364. [Google Scholar] [CrossRef]
- Krishnamoorthy, P.; Goudar, A.L.; Suresh, K.P.; Roy, P. Global and countrywide prevalence of subclinical and clinical mastitis in dairy cattle and buffaloes by systematic review and meta-analysis. Res. Vet. Sci. 2021, 136, 561–586. [Google Scholar] [CrossRef]
- Sharun, K.; Dhama, K.; Tiwari, R.; Gugjoo, M.B.; Iqbal Yatoo, M.; Patel, S.K.; Pathak, M.; Karthik, K.; Khurana, S.K.; Singh, R.; et al. Advances in therapeutic and managemental approaches of bovine mastitis: A comprehensive review. Vet. Q. 2021, 41, 107–136. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Sadiq, F.A.; Liu, T.J.; Li, Y.; Gu, J.S.; Yang, H.Y.; He, G.Q. Spoilage potential of psychrotrophic bacteria isolated from raw milk and the thermo-stability of their enzymes. J. Zhejiang Univ. Sci. B 2018, 19, 630–642. [Google Scholar] [CrossRef]
- Li, L.; Renye, J.A.; Feng, L.; Zeng, Q.; Tang, Y.; Huang, L.; Ren, D.; Yang, P. Characterization of the indigenous microflora in raw and pasteurized buffalo milk during storage at refrigeration temperature by high-throughput sequencing. J. Dairy Sci. 2016, 99, 7016–7024. [Google Scholar] [CrossRef] [PubMed]
- Éloit, M. Annual Report on Antimicrobial Agents Intended for Use in Animals, 7th ed.; World Organisation for Animals Health: Paris, France, 2023. [Google Scholar]
- Maurya, A.P.; Rajkumari, J.; Bhattacharjee, A.; Pandey, P. Development, spread and persistence of antibiotic resistance genes (ARGs) in the soil microbiomes through co-selection. Rev. Environ. Health 2020, 35, 371–378. [Google Scholar] [CrossRef]
- Thompson-Crispi, K.; Atalla, H. Bovine mastitis: Frontiers in immunogenetics. Front. Immunol. 2014, 5, 111762. [Google Scholar] [CrossRef]
- Sakwinska, O.; Bosco, N. Host microbe interactions in the lactating mammary gland. Front. Microbiol. 2019, 10, 448318. [Google Scholar] [CrossRef]
- Curone, G.; Filipe, J.; Cremonesi, P.; Trevisi, E.; Amadori, M.; Pollera, C.; Castiglioni, B.; Turin, L.; Tedde, V.; Vigo, D. What we have lost: Mastitis resistance in Holstein Friesians and in a local cattle breed. Res. Vet. Sci. 2018, 116, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Wang, Y.J.; Qin, Y.; Guix Vallverdú, R.; Maldonado García, J.; Sun, W.; Li, S.; Cao, Z. Prevalence of bovine mastitis pathogens in bulk tank milk in China. PLoS ONE 2016, 11, e0155621. [Google Scholar] [CrossRef] [PubMed]
- Yap, M.; Feehily, C.; Walsh, C.J.; Fenelon, M.; Murphy, E.F.; McAuliffe, F.M.; van Sinderen, D.; O’Toole, P.W.; O’Sullivan, O.; Cotter, P.D. Evaluation of methods for the reduction of contaminating host reads when performing shotgun metagenomic sequencing of the milk microbiome. Sci. Rep. 2020, 10, 21665. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; Pascale, G.D.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed]
- Janštová, B.; Dračková, M.; Dlesková, K.; Cupáková, Š.; Necidová, L.; Navrátilová, P.; Vorlová, L. Quality of raw milk from a farm with automatic milking system in the Czech Republic. Acta Vet. Brno 2011, 80, 207–214. [Google Scholar] [CrossRef]
- Ouamba, A.J.; Gagnon, M.; LaPointe, G.; Chouinard, P.Y.; Roy, D. Graduate Student Literature Review: Farm management practices: Potential microbial sources that determine the microbiota of raw bovine milk. J. Dairy Sci. 2022, 105, 7276–7287. [Google Scholar] [CrossRef]
- Hoque, M.N.; Istiaq, A.; Clement, R.A.; Sultana, M.; Crandall, K.A.; Siddiki, A.Z.; Hossain, M.A. Metagenomic deep sequencing reveals association of microbiome signature with functional biases in bovine mastitis. Sci. Rep. 2019, 9, 13536. [Google Scholar] [CrossRef] [PubMed]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef]
- Oniciuc, E.A.; Likotrafiti, E.; Alvarez-Molina, A.; Prieto, M.; Santos, J.A.; Alvarez-Ordóñez, A. The present and future of whole genome sequencing (WGS) and whole metagenome sequencing (WMS) for surveillance of antimicrobial resistant microorganisms and antimicrobial resistance genes across the food chain. Genes 2018, 9, 268. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Nawaz, M.; Yaqub, T.; Mushtaq, M.H. Exploring the milk microbiota of healthy and mastitic Nili Ravi buffalo using 16S rRNA gene base metagenomic analysis. Animals 2023, 13, 2298. [Google Scholar] [CrossRef] [PubMed]
- Kable, M.E.; Srisengfa, Y.; Laird, M.; Zaragoza, J.; McLeod, J.; Heidenreich, J.; Marco, M.L. The core and seasonal microbiota of raw bovine milk in tanker trucks and the impact of transfer to a milk processing facility. MBio 2016, 7, e00836-16. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, G.; Sangalli, E.; Facchi, M.; Fontana, F.; Mancabelli, L.; Donofrio, G.; Ventura, M. Metataxonomic analysis of milk microbiota in the bovine subclinical mastitis. FEMS Microbiol. Ecol. 2023, 99, fiad136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, L.; Zheng, X.; Dong, Y. Effect of yeast antagonist in combination with heat treatment on postharvest blue mold decay and Rhizopus decay of peaches. Int. J. Food Microbiol. 2007, 115, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Zheng, N.; Zhao, S.; Wang, J. Single molecule real-time sequencing and traditional cultivation techniques reveal complex community structures and regional variations of psychrotrophic bacteria in raw milk. Front. Microbiol. 2022, 13, 853263. [Google Scholar] [CrossRef] [PubMed]
- Middleton, J.R.; Fox, L.K.; Pighetti, G.; Petersson-Wolfe, C. Laboratory Handbook on Bovine Mastitis, 3rd ed.; National Mastitis Council: New Prague, MN, USA, 2017. [Google Scholar]
- Katsarou, E.I.; Katsafadou, A.I.; Karakasidis, T.; Chatzopoulos, D.C.; Vasileiou, N.G.; Lianou, D.T.; Mavrogianni, V.S.; Petinaki, E.; Fthenakis, G.C. Growth of Staphylococcus epidermidis on the surface of teatcups from milking parlours. Microorganisms 2021, 9, 852. [Google Scholar] [CrossRef]
- Sindi, A.S.; Cheema, A.S.; Trevenen, M.L.; Geddes, D.T.; Payne, M.S.; Stinson, L.F. Characterisation of human milk bacterial DNA profiles in a small cohort of Australian women in relation to infant and maternal factors. PLoS ONE 2023, 18, e0280960. [Google Scholar] [CrossRef] [PubMed]
- Mahanti, A.; Joardar, S.N.; Bandyopadhyay, S.; Banerjee, J.; Ghosh, S.; Dutta, T.K.; Samanta, I. Raw bovine milk as a reservoir of multi-drug resistant, β-lactamase-producing Klebsiella. J. Appl. Life Sci. Environ. 2024, 57, 197. [Google Scholar] [CrossRef]
- Woudstra, S.; Wente, N.; Zhang, Y.; Leimbach, S.; Kirkeby, C.; Gussmann, M.K.; Krömker, V. Reservoirs of Staphylococcus spp. and Streptococcus spp. associated with intramammary infections of dairy cows. Pathogens 2023, 12, 699. [Google Scholar] [CrossRef] [PubMed]
- Leso, L.; Barbari, M.; Lopes, M.; Damasceno, F.; Galama, P.; Taraba, J.; Kuipers, A. Invited review: Compost-bedded pack barns for dairy cows. J. Dairy Sci. 2020, 103, 1072–1099. [Google Scholar] [CrossRef]
- Hantsis-Zacharov, E.; Halpern, M. Culturable psychrotrophic bacterial communities in raw milk and their proteolytic and lipolytic traits. Appl. Environ. Microbiol. 2007, 73, 7162–7168. [Google Scholar] [CrossRef]
- Li, N.; Wang, Y.; You, C.; Ren, J.; Chen, W.; Zheng, H.; Liu, Z. Variation in raw milk microbiota throughout 12 months and the impact of weather conditions. Sci. Rep. 2018, 8, 2371. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.; Saran Netto, A.; Vaz, A.C.; Capodifóglio, E.; Gonçalves, A.; Rossi, G.A.; Figueiredo, A.S.; Ruiz, V.L. Pseudomonas spp.: Contamination sources in bulk tanks of dairy farms. Pesqui. Veterinária Bras. 2017, 37, 941–948. [Google Scholar] [CrossRef]
- Tóth, A.G.; Csabai, I.; Krikó, E.; Tőzsér, D.; Maróti, G.; Patai, Á.V.; Makrai, L.; Szita, G.; Solymosi, N. Antimicrobial resistance genes in raw milk for human consumption. Sci. Rep. 2020, 10, 7464. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Milking Method | Protein (g/100 g) | Fat (g/100 g) | Total Solid (g/100 g) | Solids-Non-Fat g/100 g) | Lactose (g/100 g) | SCC (×104/mL) | CFU (×104 CFU/mL) |
|---|---|---|---|---|---|---|---|
| HM | 4.55 ± 0.39 a | 7.33 ± 0.46 a | 17.81 ± 1.34 a | 9.77 ± 0.88 a | 4.99 ± 0.39 a | 45.85 ± 31.33 a | 37.06 ± 17.62 a |
| MM | 4.50 ± 0.29 a | 7.03 ± 0.68 a | 17.61 ± 0.76 a | 9.99 ± 0.44 a | 5.18 ± 0.20 a | 29.58 ± 15.56 a | 8.37 ± 8.37 b |
| Milking Method | Observed. | Simpson | Chao1 | ACE | Shannon | Good’s Coverage |
|---|---|---|---|---|---|---|
| HM | 830.4444 ± 49.9758 a | 0.8603 ± 0.0713 a | 852.363 ± 35.7754 a | 849.2941 ± 28.6194 a | 3.2567 ± 0.5710 a | 0.9998 ± 0.0003 a |
| MM | 665.1667 ± 71.3008 b | 0.7875 ± 0.0994 a | 777.734 ± 53.5937 b | 780.1879 ± 45.1084 b | 2.5035 ± 0.6112 b | 0.9982 ± 0.0011 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Miao, W.; Li, Z.; Huang, L.; Hau, E.; Khan, M.F.; Liu, Q.; Zeng, Q.; Cui, K. Meta-Genomic Analysis of Different Bacteria and Their Genomes Found in Raw Buffalo Milk Obtained in Various Farms Using Different Milking Methods. Genes 2024, 15, 1081. https://doi.org/10.3390/genes15081081
Li L, Miao W, Li Z, Huang L, Hau E, Khan MF, Liu Q, Zeng Q, Cui K. Meta-Genomic Analysis of Different Bacteria and Their Genomes Found in Raw Buffalo Milk Obtained in Various Farms Using Different Milking Methods. Genes. 2024; 15(8):1081. https://doi.org/10.3390/genes15081081
Chicago/Turabian StyleLi, Ling, Wenhao Miao, Zhipeng Li, Li Huang, Enghuan Hau, Muhammad Farhan Khan, Qingyou Liu, Qingkun Zeng, and Kuiqing Cui. 2024. "Meta-Genomic Analysis of Different Bacteria and Their Genomes Found in Raw Buffalo Milk Obtained in Various Farms Using Different Milking Methods" Genes 15, no. 8: 1081. https://doi.org/10.3390/genes15081081