Genetic and Pathophysiological Basis of Cardiac and Skeletal Muscle Laminopathies


Abstract
:1. Introduction
Intermediate Filaments, Lamins Types, and Function
2. Laminopathies (One Gene Multiple Pathologies)
3. Striated Muscle Laminopathy

4. Modeling Striated Muscle Laminopathies
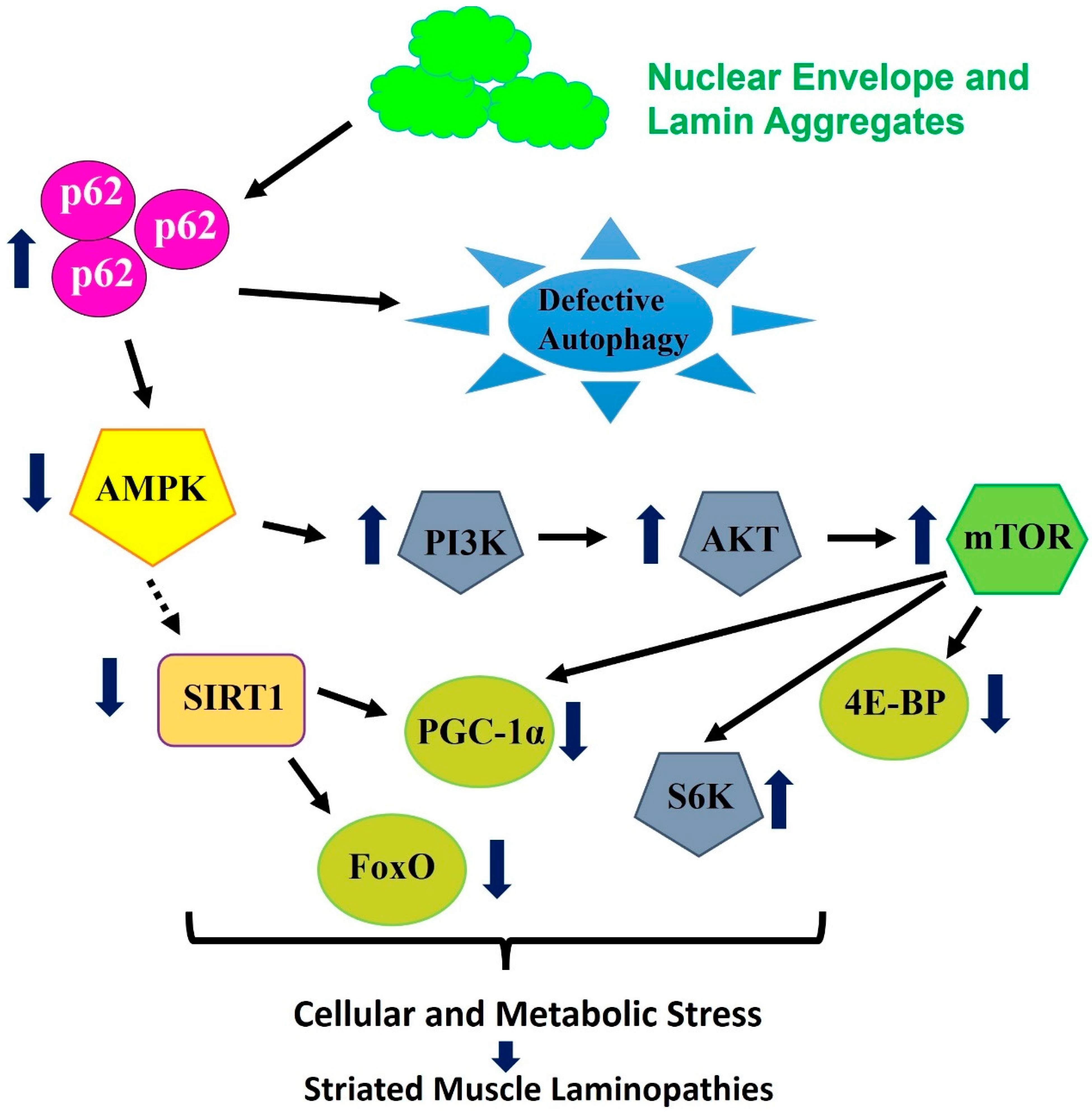
5. Pathways and Signaling Associated with Striated Muscle Laminopathies and Aging
6. Redox Signaling and Autophagy Association with Laminopathies
7. Proteostasis Signaling with Cytoplasmic-Linked Aggregation Laminopathies
8. Treatments and Future Prospective
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NPC | Nuclear Pore Complex |
| NEP | Nuclear envelope proteins |
| LINC | Linker of nucleoskeleton and cytoskeleton |
| EDMD | Emery–Dreifuss muscular dystrophy |
| HGPS | Hutchinson–Gilford progeria syndrome |
| LGMD | Limb–girdle muscular dystrophy |
| FPLD | Familial partial lipodystrophy of the Dunnigan type |
| DCM | Dilated cardiomyopathy |
| CMD | Congenital muscular dystrophy |
References
- Dechat, T.; Adam, S.A.; Goldman, R.D. Nuclear lamins and chromatin: When structure meets function. Adv. Enzym. Regul. 2009, 49, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.D.; Gruenbaum, Y.; Moir, R.D.; Shumaker, D.K.; Spann, T.P. Nuclear lamins: Building blocks of nuclear architecture. Genes Dev. 2002, 16, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Bar, H.; Kreplak, L.; Strelkov, S.V.; Aebi, U. Intermediate filaments: From cell architecture to nanomechanics. Nat. Rev. Mol. Cell Biol. 2007, 8, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Hurt, E. The nuclear pore complex: Understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 2017, 18, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.A.; Goldman, R.D. Insights into the differences between the A- and B-type nuclear lamins. Adv. Biol. Regul. 2012, 52, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Stick, R. The gene structure of B-type nuclear lamins of Xenopus laevis: Implications for the evolution of the vertebrate lamin family. Chromosome Res. 1994, 2, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Melkani, G.C.; Bernstein, S.I. The UNC-45 myosin chaperone: From worms to flies to vertebrates. Int. Rev. Cell Mol. Biol. 2014, 313, 103–144. [Google Scholar] [CrossRef] [PubMed]
- Camps, J.; Erdos, M.R.; Ried, T. The role of lamin B1 for the maintenance of nuclear structure and function. Nucleus 2015, 6, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Young, S.G.; Jung, H.J.; Lee, J.M.; Fong, L.G. Nuclear lamins and neurobiology. Mol. Cell. Biol. 2014, 34, 2776–2785. [Google Scholar] [CrossRef] [PubMed]
- Broers, J.L.; Ramaekers, F.C.; Bonne, G.; Yaou, R.B.; Hutchison, C.J. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol. Rev. 2006, 86, 967–1008. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Adam, S.A.; Taimen, P.; Shimi, T.; Goldman, R.D. Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000547. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, T.A.; Misteli, T. The lamin protein family. Genome Biol. 2011, 12, 222. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Alsheimer, M.; von Glasenapp, E.; Schnolzer, M.; Heid, H.; Benavente, R. Meiotic lamin C2: The unique amino-terminal hexapeptide GNAEGR is essential for nuclear envelope association. Proc. Natl. Acad. Sci. USA 2000, 97, 13120–13125. [Google Scholar] [CrossRef] [PubMed]
- Machiels, B.M.; Zorenc, A.H.; Endert, J.M.; Kuijpers, H.J.; van Eys, G.J.; Ramaekers, F.C.; Broers, J.L. An alternative splicing product of the lamin A/C gene lacks exon 10. J. Biol. Chem. 1996, 271, 9249–9253. [Google Scholar] [CrossRef] [PubMed]
- Moir, R.D.; Spann, T.P.; Lopez-Soler, R.I.; Yoon, M.; Goldman, A.E.; Khuon, S.; Goldman, R.D. Review: The dynamics of the nuclear lamins during the cell cycle—Relationship between structure and function. J. Struct. Biol. 2000, 129, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Somovilla-Crespo, B.; Rius, C.; Gonzalez-Granado, J.M. Lamin A/C and the Immune System: One Intermediate Filament, Many Faces. Int. J. Mol. Sci. 2020, 21, 6109. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, K.; Pelletier, M.G.H.; Malu, K.; Barbeau, A.M.; Giadone, R.M.; Babroudi, S.C.; Gaines, P.C.W. Expression Levels of Lamin A or C Are Critical to Nuclear Maturation, Functional Responses, and Gene Expression Profiles in Differentiating Mouse Neutrophils. Immunohorizons 2022, 6, 16–35. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.D.; Grin, B.; Mendez, M.G.; Kuczmarski, E.R. Intermediate filaments: Versatile building blocks of cell structure. Curr. Opin. Cell Biol. 2008, 20, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Mounkes, L.C.; Kozlov, S.V.; Rottman, J.N.; Stewart, C.L. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum. Mol. Genet. 2005, 14, 2167–2180. [Google Scholar] [CrossRef] [PubMed]
- Lenz-Bohme, B.; Wismar, J.; Fuchs, S.; Reifegerste, R.; Buchner, E.; Betz, H.; Schmitt, B. Insertional mutation of the Drosophila nuclear lamin Dm0 gene results in defective nuclear envelopes, clustering of nuclear pore complexes, and accumulation of annulate lamellae. J. Cell Biol. 1997, 137, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, K.; Williams, T.; Krasnow, M.A. A nuclear lamin is required for cytoplasmic organization and egg polarity in Drosophila. Nat. Cell Biol. 2001, 3, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Soler, R.I.; Moir, R.D.; Spann, T.P.; Stick, R.; Goldman, R.D. A role for nuclear lamins in nuclear envelope assembly. J. Cell Biol. 2001, 154, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rolef Ben-Shahar, T.; Riemer, D.; Treinin, M.; Spann, P.; Weber, K.; Fire, A.; Gruenbaum, Y. Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression, and spatial organization of nuclear pore complexes. Mol. Biol. Cell 2000, 11, 3937–3947. [Google Scholar] [CrossRef] [PubMed]
- van Engelen, B.G.; Muchir, A.; Hutchison, C.J.; van der Kooi, A.J.; Bonne, G.; Lammens, M. The lethal phenotype of a homozygous nonsense mutation in the lamin A/C gene. Neurology 2005, 64, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, V.; Leimena, C.; McMahon, A.C.; Tan, J.C.; Chandar, S.; Jogia, D.; Kesteven, S.H.; Michalicek, J.; Otway, R.; Verheyen, F.; et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Investig. 2004, 113, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.J.; Chen, S.C.; Garelick, M.G.; Dai, D.F.; Liao, C.Y.; Schreiber, K.H.; MacKay, V.L.; An, E.H.; Strong, R.; Ladiges, W.C.; et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012, 4, 144ra03. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.; Roux, K.J. Nuclei take a position: Managing nuclear location. Dev. Cell 2009, 17, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Hutchinson, P.J.; Hale, C.M.; Wirtz, D.; Hodzic, D. Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Exp. Cell Res. 2008, 314, 1892–1905. [Google Scholar] [CrossRef] [PubMed]
- Vigouroux, C.; Bonne, G. Laminopathies: One Gene, Two Proteins, Five Diseases. In Nuclear Envelope Dynamics in Embryos and Somatic Cells; Landes Bioscience: Georgetown, TX, USA, 2002; pp. 153–172. [Google Scholar]
- Davidson, P.M.; Lammerding, J. Broken nuclei—Lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014, 24, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Lammerding, J. Lamins at a glance. J. Cell Sci. 2012, 125 Pt 9, 2087–2093. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Fong, L.G.; Muchir, A.; Young, S.G. Laminopathies and the long strange trip from basic cell biology to therapy. J. Clin. Investig. 2009, 119, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Crasto, S.; My, I.; Di Pasquale, E. The Broad Spectrum of LMNA Cardiac Diseases: From Molecular Mechanisms to Clinical Phenotype. Front. Physiol. 2020, 11, 761. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pombo, A.; Diaz-Lopez, E.J.; Castro, A.I.; Sanchez-Iglesias, S.; Cobelo-Gomez, S.; Prado-Morana, T.; Araujo-Vilar, D. Clinical Spectrum of LMNA-Associated Type 2 Familial Partial Lipodystrophy: A Systematic Review. Cells 2023, 12, 725. [Google Scholar] [CrossRef] [PubMed]
- Dutour, A.; Roll, P.; Gaborit, B.; Courrier, S.; Alessi, M.C.; Tregouet, D.A.; Angelis, F.; Robaglia-Schlupp, A.; Lesavre, N.; Cau, P.; et al. High prevalence of laminopathies among patients with metabolic syndrome. Hum. Mol. Genet. 2011, 20, 3779–3786. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Columbaro, M.; Schena, E.; Prencipe, S.; Andrenacci, D.; Iozzo, P.; Angela Guzzardi, M.; Capanni, C.; Mattioli, E.; Loi, M.; et al. Altered adipocyte differentiation and unbalanced autophagy in type 2 Familial Partial Lipodystrophy: An in vitro and in vivo study of adipose tissue browning. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Carboni, N.; Bernasconi, P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells 2016, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zabell, A.; Koh, W.; Tang, W.H. Lamin A/C Cardiomyopathies: Current Understanding and Novel Treatment Strategies. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- Azibani, F.; Muchir, A.; Vignier, N.; Bonne, G.; Bertrand, A.T. Striated muscle laminopathies. Semin. Cell Dev. Biol. 2014, 29, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, G.; Benedetti, S.; Bertini, E.; Boriani, G.; Mazzanti, L.; Novelli, G.; Pasquali, R.; Pini, A.; Politano, L. Laminopathies: Many diseases, one gene. Report of the first Italian Meeting Course on Laminopathies. Acta Myol. 2011, 30, 138–143. [Google Scholar] [PubMed]
- Decostre, V.; Ben Yaou, R.; Bonne, G. Laminopathies affecting skeletal and cardiac muscles: Clinical and pathophysiological aspects. Acta Myol. 2005, 24, 104–109. [Google Scholar] [PubMed]
- Butin-Israeli, V.; Adam, S.A.; Goldman, A.E.; Goldman, R.D. Nuclear lamin functions and disease. Trends Genet. 2012, 28, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Carboni, N.; Mateddu, A.; Marrosu, G.; Cocco, E.; Marrosu, M.G. Genetic and clinical characteristics of skeletal and cardiac muscle in patients with lamin A/C gene mutations. Muscle Nerve 2013, 48, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Charniot, J.C.; Desnos, M.; Zerhouni, K.; Bonnefont-Rousselot, D.; Albertini, J.P.; Salama, J.Z.; Bassez, G.; Komajda, M.; Artigou, J.Y. Severe dilated cardiomyopathy and quadriceps myopathy due to lamin A/C gene mutation: A phenotypic study. Eur. J. Heart Fail. 2006, 8, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Ausma, J.; van Eys, G.J.; Broers, J.L.; Thone, F.; Flameng, W.; Ramaekers, F.C.; Borgers, M. Nuclear lamin expression in chronic hibernating myocardium in man. J. Mol. Cell. Cardiol. 1996, 28, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Goidescu, C.M. Dilated cardiomyopathy produced by lamin A/C gene mutations. Clujul Med. 2013, 86, 309–312. [Google Scholar] [PubMed]
- Orie, J.E.; Liedtke, A.J. Cardiomyopathy. 2. Hypertrophic and restrictive/obliterative types. Postgrad. Med. 1986, 79, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Pilotto, A.; Repetto, A.; Grasso, M.; Negri, A.; Diegoli, M.; Campana, C.; Scelsi, L.; Baldini, E.; Gavazzi, A.; et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: A lamin A/C defect-related disease. J. Am. Coll. Cardiol. 2002, 39, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Bilinska, Z.T.; Sylvius, N.; Grzybowski, J.; Fidzianska, A.; Michalak, E.; Walczak, E.; Walski, M.; Bieganowska, K.; Szymaniak, E.; Kusmierczyk-Droszcz, B.; et al. Dilated cardiomyopathy caused by LMNA mutations. Clinical and morphological studies. Kardiol. Pol. 2006, 64, 812–819, discussion 20-1. [Google Scholar] [PubMed]
- Carboni, N.; Sardu, C.; Cocco, E.; Marrosu, G.; Manzi, R.C.; Nissardi, V.; Isola, F.; Mateddu, A.; Solla, E.; Maioli, M.A.; et al. Cardiac involvement in patients with lamin A/C gene mutations: A cohort observation. Muscle Nerve 2012, 46, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Bhide, S.; Trujillo, A.S.; O’Connor, M.T.; Young, G.H.; Cryderman, D.E.; Chandran, S.; Nikravesh, M.; Wallrath, L.L.; Melkani, G.C. Increasing autophagy and blocking Nrf2 suppress laminopathy-induced age-dependent cardiac dysfunction and shortened lifespan. Aging Cell 2018, 17, e12747. [Google Scholar] [CrossRef] [PubMed]
- Chandar, S.; Yeo, L.S.; Leimena, C.; Tan, J.C.; Xiao, X.H.; Nikolova-Krstevski, V.; Yasuoka, Y.; Gardiner-Garden, M.; Wu, J.; Kesteven, S.; et al. Effects of mechanical stress and carvedilol in lamin A/C-deficient dilated cardiomyopathy. Circ. Res. 2010, 106, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Ikram, S.; Bibi, N.; Mir, A. Hutchinson-Gilford Progeria Syndrome: A Premature Aging Disease. Mol. Neurobiol. 2018, 55, 4417–4427. [Google Scholar] [CrossRef] [PubMed]
- Apte, K.; Stick, R.; Radmacher, M. Mechanics in human fibroblasts and progeria: Lamin A mutation E145K results in stiffening of nuclei. J. Mol. Recognit. 2017, 30, e2580. [Google Scholar] [CrossRef] [PubMed]
- Cenni, V.; D’Apice, M.R.; Garagnani, P.; Columbaro, M.; Novelli, G.; Franceschi, C.; Lattanzi, G. Mandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing. Ageing Res. Rev. 2018, 42, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.; Suggs, J.A.; Wang, B.J.; Han, A.; Bhide, S.; Cryderman, D.E.; Moore, S.A.; Bernstein, S.I.; Wallrath, L.L.; Melkani, G.C. Suppression of myopathic lamin mutations by muscle-specific activation of AMPK and modulation of downstream signaling. Hum. Mol. Genet. 2019, 28, 351–371. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; van Engelen, B.G.; Lammens, M.; Mislow, J.M.; McNally, E.; Schwartz, K.; Bonne, G. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp. Cell Res. 2003, 291, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.P.; van den Berg, C.W.; Casini, S.; Braam, S.R.; Mummery, C.L. Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends Mol. Med. 2011, 17, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Evans, T. Embryonic Stem Cells as a Model for Cardiac Development and Disease. Drug Discov. Today Dis. Models 2008, 5, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Lau, Y.M.; Cai, Z.J.; Lai, W.H.; Wong, L.Y.; Tse, H.F.; Ng, K.M.; Siu, C.W. Modeling Treatment Response for Lamin A/C Related Dilated Cardiomyopathy in Human Induced Pluripotent Stem Cells. J. Am. Heart Assoc. 2017, 6, e005677. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Suzuki, K.; Qu, J.; Sancho-Martinez, I.; Yi, F.; Li, M.; Kumar, S.; Nivet, E.; Kim, J.; Soligalla, R.D.; et al. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell 2011, 8, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Bank, E.M.; Ben-Harush, K.; Wiesel-Motiuk, N.; Barkan, R.; Feinstein, N.; Lotan, O.; Medalia, O.; Gruenbaum, Y. A laminopathic mutation disrupting lamin filament assembly causes disease-like phenotypes in Caenorhabditis elegans. Mol. Biol. Cell 2011, 22, 2716–2728. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.H.; Kennedy, B.K. When lamins go bad: Nuclear structure and disease. Cell 2013, 152, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Koshimizu, E.; Imamura, S.; Qi, J.; Toure, J.; Valdez, D.M., Jr.; Carr, C.E.; Hanai, J.; Kishi, S. Embryonic senescence and laminopathies in a progeroid zebrafish model. PLoS ONE 2011, 6, e17688. [Google Scholar] [CrossRef] [PubMed]
- Schulze, S.R.; Curio-Penny, B.; Li, Y.; Imani, R.A.; Rydberg, L.; Geyer, P.K.; Wallrath, L.L. Molecular genetic analysis of the nested Drosophila melanogaster lamin C gene. Genetics 2005, 171, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Palka, M.; Tomczak, A.; Grabowska, K.; Machowska, M.; Piekarowicz, K.; Rzepecka, D.; Rzepecki, R. Laminopathies: What can humans learn from fruit flies. Cell. Mol. Biol. Lett. 2018, 23, 32. [Google Scholar] [CrossRef] [PubMed]
- Shevelyov, Y.Y.; Lavrov, S.A.; Mikhaylova, L.M.; Nurminsky, I.D.; Kulathinal, R.J.; Egorova, K.S.; Rozovsky, Y.M.; Nurminsky, D.I. The B-type lamin is required for somatic repression of testis-specific gene clusters. Proc. Natl. Acad. Sci. USA 2009, 106, 3282–3287. [Google Scholar] [CrossRef] [PubMed]
- Osouda, S.; Nakamura, Y.; de Saint Phalle, B.; McConnell, M.; Horigome, T.; Sugiyama, S.; Fisher, P.A.; Furukawa, K. Null mutants of Drosophila B-type lamin Dm(0) show aberrant tissue differentiation rather than obvious nuclear shape distortion or specific defects during cell proliferation. Dev. Biol. 2005, 284, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Gurudatta, B.V.; Shashidhara, L.S.; Parnaik, V.K. Lamin C and chromatin organization in Drosophila. J. Genet. 2010, 89, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Dialynas, G.; Speese, S.; Budnik, V.; Geyer, P.K.; Wallrath, L.L. The role of Drosophila Lamin C in muscle function and gene expression. Development 2010, 137, 3067–3077. [Google Scholar] [CrossRef] [PubMed]
- Dialynas, G.; Flannery, K.M.; Zirbel, L.N.; Nagy, P.L.; Mathews, K.D.; Moore, S.A.; Wallrath, L.L. LMNA variants cause cytoplasmic distribution of nuclear pore proteins in Drosophila and human muscle. Hum. Mol. Genet. 2012, 21, 1544–1556. [Google Scholar] [CrossRef] [PubMed]
- Dialynas, G.; Shrestha, O.K.; Ponce, J.M.; Zwerger, M.; Thiemann, D.A.; Young, G.H.; Moore, S.A.; Yu, L.; Lammerding, J.; Wallrath, L.L. Myopathic lamin mutations cause reductive stress and activate the nrf2/keap-1 pathway. PLoS Genet. 2015, 11, e1005231. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hassinger, L.; Thomson, T.; Ding, B.; Ashley, J.; Hassinger, W.; Budnik, V. Lamin Mutations Accelerate Aging via Defective Export of Mitochondrial mRNAs through Nuclear Envelope Budding. Curr. Biol. 2016, 26, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Jung, H.J.; Nobumori, C.; Chang, S.; Tu, Y.; Barnes, R.H., 2nd; Yoshinaga, Y.; de Jong, P.J.; Vergnes, L.; Reue, K.; et al. Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol. Biol. Cell 2011, 22, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Chang, S.Y.; Yin, L.; Tu, Y.; Hu, Y.; Yoshinaga, Y.; de Jong, P.J.; Fong, L.G.; Young, S.G. An absence of both lamin B1 and lamin B2 in keratinocytes has no effect on cell proliferation or the development of skin and hair. Hum. Mol. Genet. 2011, 20, 3537–3544. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Helbling-Leclerc, A.; Massart, C.; Varnous, S.; Niel, F.; Lacene, E.; Fromes, Y.; Toussaint, M.; Mura, A.M.; Keller, D.I.; et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 2005, 14, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.T.; Renou, L.; Papadopoulos, A.; Beuvin, M.; Lacene, E.; Massart, C.; Ottolenghi, C.; Decostre, V.; Maron, S.; Schlossarek, S.; et al. DelK32-lamin A/C has abnormal location and induces incomplete tissue maturation and severe metabolic defects leading to premature death. Hum. Mol. Genet. 2012, 21, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Voncken, J.W.; Konings, G.; van Weeghel, M.; van den Hoogenhof, M.M.; Gijbels, M.; van Erk, A.; Schoonderwoerd, K.; van den Bosch, B.; Dahlmans, V.; et al. Post-natal myogenic and adipogenic developmental: Defects and metabolic impairment upon loss of A-type lamins. Nucleus 2011, 2, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Li, W.; Vidal, C.; Yeo, L.S.; Fatkin, D.; Duque, G. Lamin A/C deficiency is associated with fat infiltration of muscle and bone. Mech. Ageing Dev. 2011, 132, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.L.; Kozlov, S.; Fong, L.G.; Young, S.G. Mouse models of the laminopathies. Exp. Cell Res. 2007, 313, 2144–2156. [Google Scholar] [CrossRef] [PubMed]
- Mounkes, L.C.; Kozlov, S.; Hernandez, L.; Sullivan, T.; Stewart, C.L. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 2003, 423, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Morales, A.; Siegfried, J.D. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet. Med. 2010, 12, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Vaikhanskaya, T.; Sivitskaya, L.; Danilenko, N.; Davydenko, O.; Kurushka, T.; Sidorenko, I. LMNA-related dilated cardiomyopathy. Oxf. Med. Case Rep. 2014, 2014, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.C.; Worman, H.J. Reactivation of autophagy ameliorates LMNA cardiomyopathy. Autophagy 2013, 9, 110–111. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.H.; Misteli, T. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Wallrath, L.L.; Bohnekamp, J.; Magin, T.M. Cross talk between the cytoplasm and nucleus during development and disease. Curr. Opin. Genet. Dev. 2016, 37, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; deBeer, J.; Tarnopolsky, M.A. Dysfunctional Nrf2-Keap1 redox signaling in skeletal muscle of the sedentary old. Free Radic. Biol. Med. 2010, 49, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Stepkowski, T.M.; Kruszewski, M.K. Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radic. Biol. Med. 2011, 50, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, B.J.; Isakson, P.; Lewerenz, J.; Sanchez, H.; Kotzebue, R.W.; Cumming, R.C.; Harris, G.L.; Nezis, I.P.; Schubert, D.R.; Simonsen, A.; et al. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 2011, 7, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Nezis, I.P.; Stenmark, H. p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxid. Redox Signal. 2012, 17, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.C.; Muchir, A.; Wu, W.; Iwata, S.; Homma, S.; Morrow, J.P.; Worman, H.J. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 2012, 4, 144ra02. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T. Feedback on fat: p62-mTORC1-autophagy connections. Cell 2011, 147, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.E.; Hayashi, Y.K.; Goto, K.; Komaki, H.; Hayashi, Y.; Inuzuka, T.; Noguchi, S.; Nonaka, I.; Nishino, I. Nuclear changes in skeletal muscle extend to satellite cells in autosomal dominant Emery-Dreifuss muscular dystrophy/limb-girdle muscular dystrophy 1B. Neuromuscul Disord. 2009, 19, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Egesipe, A.L.; Blondel, S.; Lo Cicero, A.; Jaskowiak, A.L.; Navarro, C.; Sandre-Giovannoli, A.; Levy, N.; Peschanski, M.; Nissan, X. Metformin decreases progerin expression and alleviates pathological defects of Hutchinson-Gilford progeria syndrome cells. NPJ Aging Mech. Dis. 2016, 2, 16026. [Google Scholar] [CrossRef] [PubMed]
- Demontis, F.; Perrimon, N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell 2010, 143, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Zid, B.M.; Rogers, A.N.; Katewa, S.D.; Vargas, M.A.; Kolipinski, M.C.; Lu, T.A.; Benzer, S.; Kapahi, P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 2009, 139, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.Y.; Anderson, S.S.; Chicoine, N.H.; Mayfield, J.R.; Garrett, B.J.; Kwok, C.S.; Academia, E.C.; Hsu, Y.M.; Miller, D.M.; Bair, A.M.; et al. Evidence that S6K1, but not 4E-BP1, mediates skeletal muscle pathology associated with loss of A-type lamins. Cell Discov. 2017, 3, 17039. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Amendola, M.; van Steensel, B. Mechanisms and dynamics of nuclear lamina-genome interactions. Curr. Opin. Cell Biol. 2014, 28, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Gnocchi, V.F.; Scharner, J.; Huang, Z.; Brady, K.; Lee, J.S.; White, R.B.; Morgan, J.E.; Sun, Y.B.; Ellis, J.A.; Zammit, P.S. Uncoordinated transcription and compromised muscle function in the lmna-null mouse model of Emery- Emery-Dreyfuss muscular dystrophy. PLoS ONE 2011, 6, e16651. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Junger, M.A.; Rintelen, F.; Stocker, H.; Wasserman, J.D.; Vegh, M.; Radimerski, T.; Greenberg, M.E.; Hafen, E. The Drosophila forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J. Biol. 2003, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhang, Z.; Saha, A.; Peng, S.; Chandra, G.; Quezado, Z.; Mukherjee, A.B. Disruption of adaptive energy metabolism and elevated ribosomal p-S6K1 levels contribute to INCL pathogenesis: Partial rescue by resveratrol. Hum. Mol. Genet. 2011, 20, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Teleman, A.A.; Chen, Y.W.; Cohen, S.M. 4E-BP functions as a metabolic brake used under stress conditions but not during normal growth. Genes Dev. 2005, 19, 1844–1848. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Wang, L.; Zhang, J.; Numazawa, S.; Tang, H.; Tang, X.; Han, X.; Li, J.; Yang, M.; Wang, Z.; et al. The crosstalk between Nrf2 and AMPK signal pathways is important for the anti-inflammatory effect of berberine in LPS-stimulated macrophages and endotoxin-shocked mice. Antioxid. Redox Signal. 2014, 20, 574–588. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| LMNA/LamC (Human/Drosophila) | Observed Striated Muscle Phenotype/Life Span in Mouse or Drosophila Models (Only Human Amino Acids LMNA Mutations Drosophila Orthologs Are Shown) |
|---|---|
| Lmna–/–) | Compromised cardiac and skeletal muscle performance and short lifespan. |
| Lmna-E159K | Swollen mitochondria, defective proteostasis, enhanced ROS, and myofibrillar disorganization. |
| Lmna-L162P | Progressive muscle dysfunction, shuttle nuclear blebbing, and LamC aggregates. |
| Lmna-R190W | Dilated cardiomyopathy, muscle dysfunction, nuclear blebbing, LamC aggregates, short lifespan, defective autophagy, and redox signaling. |
| Lmna-N195K | Mislocation of transcription factor Hf1b/Sp4, loss of sarcomere function, reduced lifespan to arrhythmia. |
| Lmna-H222P | Muscular dystrophies and dilated cardiomyopathy. |
| Lmna-M371K | Nuclear defects, abnormal heart pathology, and short lifespan. |
| LamC-G449V | Constricted heart, muscle dysfunction, nuclear blebbing, cytoplasmic LamC aggregates, short lifespan, defective autophagy, and redox signaling. |
| LamC-L489P | Severe skeletal myopathy with nuclear blebbing, and cytoplasmic LamC aggregates. |
| LMNA-L530P | The homozygous mutant mice have significantly compromised growth and all the mice were not viable after a month. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhide, S.; Chandran, S.; Rajasekaran, N.S.; Melkani, G.C. Genetic and Pathophysiological Basis of Cardiac and Skeletal Muscle Laminopathies. Genes 2024, 15, 1095. https://doi.org/10.3390/genes15081095
Bhide S, Chandran S, Rajasekaran NS, Melkani GC. Genetic and Pathophysiological Basis of Cardiac and Skeletal Muscle Laminopathies. Genes. 2024; 15(8):1095. https://doi.org/10.3390/genes15081095
Chicago/Turabian StyleBhide, Shruti, Sahaana Chandran, Namakkal S. Rajasekaran, and Girish C. Melkani. 2024. "Genetic and Pathophysiological Basis of Cardiac and Skeletal Muscle Laminopathies" Genes 15, no. 8: 1095. https://doi.org/10.3390/genes15081095