
Fine Mapping and Candidate Gene Analysis of Two Major Quantitative Trait Loci, qFW2.1 and qFW3.1, Controlling Fruit Weight in Pepper (Capsicum annuum)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction, PCR Amplification, and Detection
2.3. QTL-Seq Analysis
2.4. QTL Mapping with InDel Marker
2.5. Transcriptome Analysis
2.6. Verification of Gene Expression Pattern Using qRT-PCR
3. Results
3.1. Phenotypic Analysis of F2
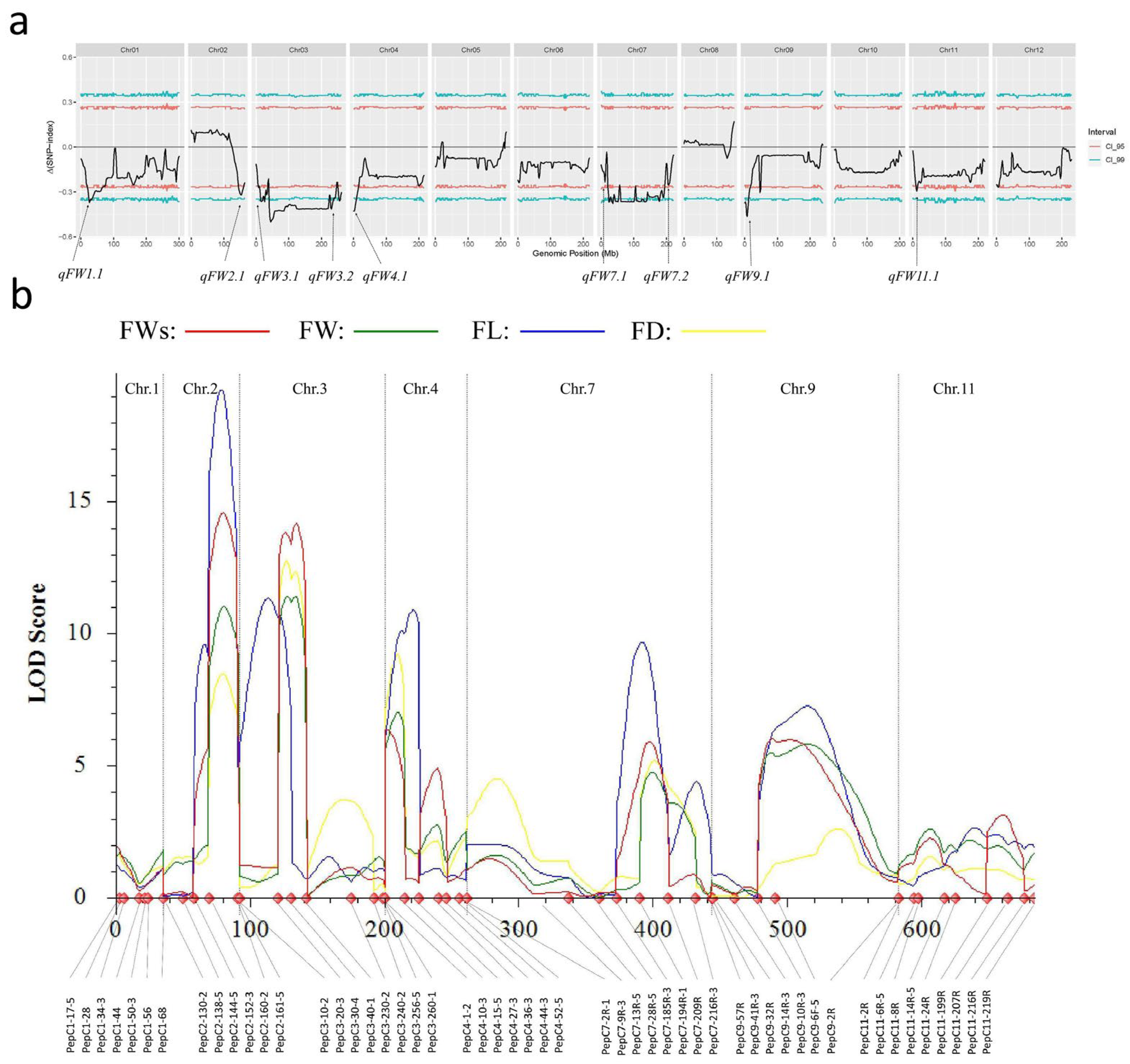
3.2. Two Major QTL Were Identified by Using QTL-Seq
3.3. Genetic Linkage Analysis Narrowed qFW2.1 and qFW3.1 Candidate Region
3.4. Differential Gene Expression Analysis between the Two Parents
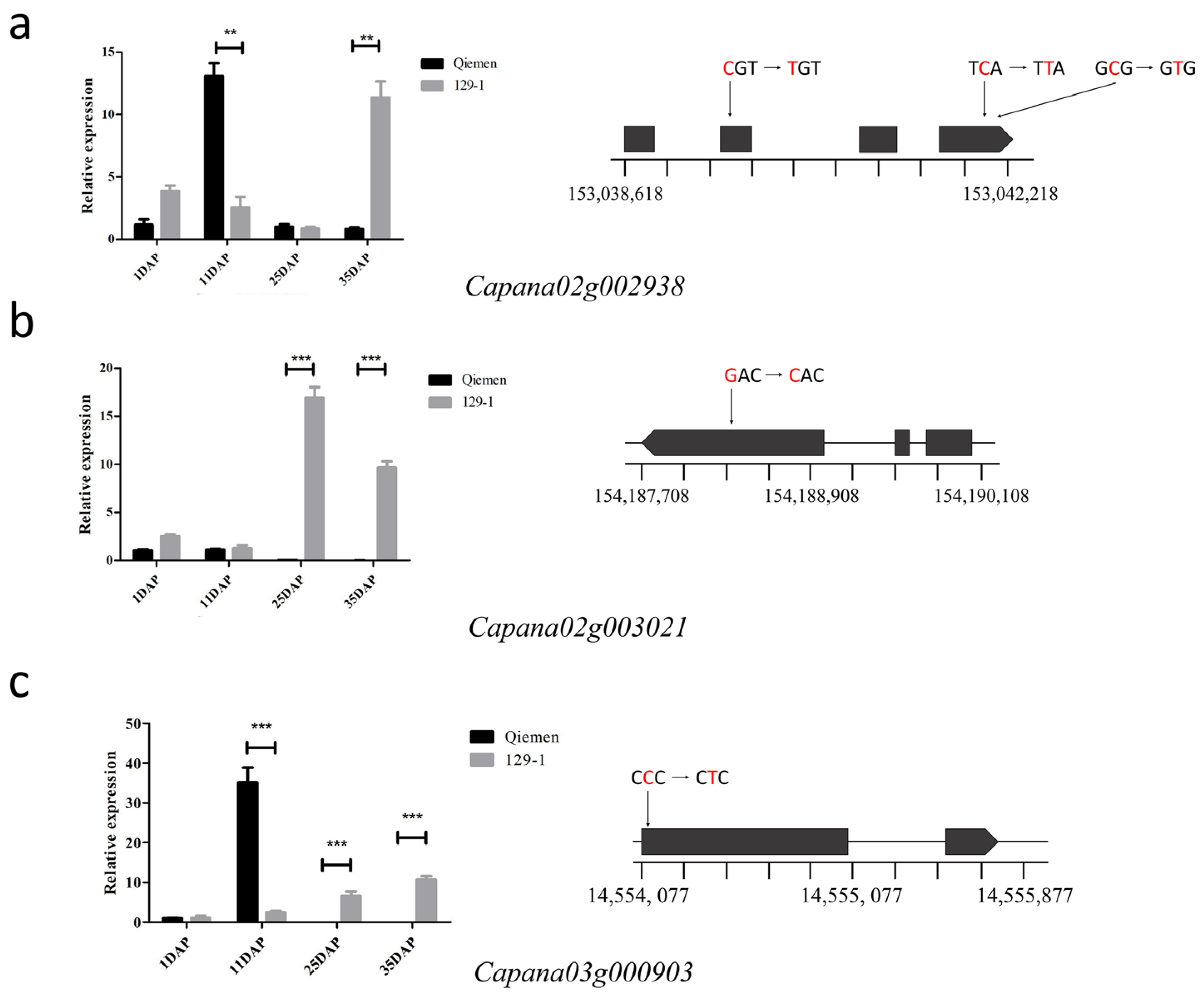
3.5. Prediction and Analysis of Candidate Genes for the Fruit Weight
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barboza, G.E.; García, C.C.; de Bem Bianchetti, L.; Romero, M.V.; Scaldaferro, M. Monograph of wild and cultivated chili peppers (Capsicum L., Solanaceae). PhytoKeys 2022, 200, 1. [Google Scholar]
- Baenas, N.; Belović, M.; Ilic, N.; Moreno, D.A.; García-Viguera, C. Industrial use of pepper (Capsicum annum L.) derived products: Technological benefits and biological advantages. Food Chem. 2019, 274, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, R.N.; Koshima, C.C.; Forster-Carneiro, T.; Gomes, M.T.M.S.; Rostagno, M.A.; Prado, J.M.; Meireles, M.A.A. Uses and applications of extracts from natural sources. In Natural Product Extraction: Principles and Applications; Royal Society of Chemistry: London, UK, 2022. [Google Scholar]
- Sinclair, T.R.; Purcell, L.C.; Sneller, C.H. Crop transformation and the challenge to increase yield potential. Trends Plant Sci. 2004, 9, 70–75. [Google Scholar] [CrossRef]
- Ariizumi, T.; Shinozaki, Y.; Ezura, H. Genes that influence yield in tomato. Breed. Sci. 2013, 63, 3–13. [Google Scholar] [CrossRef]
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The Molecular Genetics of Crop Domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef]
- Paran, I.; Van Der Knaap, E. Genetic and molecular regulation of fruit and plant domestication traits in tomato and pepper. J. Exp. Bot. 2007, 58, 3841–3852. [Google Scholar] [CrossRef] [PubMed]
- Seymour, G.B.; Chapman, N.H.; Chew, B.L.; Rose, J.K. Regulation of ripening and opportunities for control in tomato and other fruits. Plant Biotechnol. J. 2013, 11, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Frary, A.; Nesbitt, T.C.; Grandillo, S.; Knaap, E.; Cong, B.; Liu, J.; Meller, J.; Elber, R.; Alpert, K.B.; Tanksley, S.D. fw2.2: A Quantitative Trait Locus Key to the Evolution of Tomato Fruit Size. Science 2000, 289, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Zhang, N.; Sauvage, C.; Muños, S.; Blanca, J.; Cañizares, J.; Diez, M.J.; Schneider, R.; Mazourek, M.; McClead, J.; et al. A cytochrome P450 regulates a domestication trait in cultivated tomato. Proc. Natl. Acad. Sci. USA 2013, 110, 17125–17130. [Google Scholar] [CrossRef]
- Mu, Q.; Huang, Z.; Chakrabarti, M.; Illa-Berenguer, E.; Liu, X.; Wang, Y.; Ramos, A.; van der Knaap, E. Fruit weight is controlled by Cell Size Regulator encoding a novel protein that is expressed in maturing tomato fruits. PLoS Genet. 2017, 13, e1006930. [Google Scholar] [CrossRef]
- Rodriguez, G.R.; Muños, S.; Anderson, C.; Sim, S.C.; Michel, A.; Causse, M.; McSpadden Gardener, B.B.; Francis, D.; van Der Knaap, E. Distribution of SUN, OVATE, LC, and FAS in the tomato germplasm and the relationship to fruit shape diversity. Plant Physiol. 2011, 156, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Muños, S.; Ranc, N.; Botton, E.; Bérard, A.; Rolland, S.; Duffé, P.; Carretero, Y.; Le Paslier, M.-C.; Delalande, C.; Bouzayen, M.; et al. Increase in Tomato Locule Number Is Controlled by Two Single-Nucleotide Polymorphisms Located Near WUSCHEL. Plant Physiol. 2011, 156, 2244–2254. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Orozco, E.; Shekasteband, R.; Illa-Berenguer, E.; Snouffer, A.; van der Knaap, E.; Lee, T.G.; Hutton, S.F. Identification and characterization of GLOBE, a major gene controlling fruit shape and impacting fruit size and marketability in tomato. Hortic. Res. 2021, 8, 138. [Google Scholar] [CrossRef]
- Penchovsky, R.; Kaloudas, D. Molecular factors affecting tomato fruit size. Plant Gene 2023, 33, 100395. [Google Scholar] [CrossRef]
- Borovsky, Y.; Doron-Faigenboim, A.; Paran, I. Pepper fruit elongation is controlled by Capsicum annuum ovate family protein 20. Front. Plant Sci. 2022, 12, 815589. [Google Scholar] [CrossRef]
- Chaim, A.B.; Paran, I.; Grube, R.C.; Jahn, M.; Van Wijk, R.; Peleman, J. QTL mapping of fruit-related traits in pepper (Capsicum annuum). Theor. Appl. Genet. 2001, 102, 1016–1028. [Google Scholar] [CrossRef]
- Zygier, S.; Chaim, A.B.; Efrati, A.; Kaluzky, G.; Borovsky, Y.; Paran, I. QTLs mapping for fruit size and shape in chromosomes 2 and 4 in pepper and a comparison of the pepper QTL map with that of tomato. Theor. Appl. Genet. 2005, 111, 437–445. [Google Scholar] [CrossRef]
- Rao, G.U.; Ben Chaim, A.; Borovsky, Y.; Paran, I. Mapping of yield-related QTLs in pepper in an interspecific cross of Capsicum annuum and C. frutescens. Theor. Appl. Genet. 2003, 106, 1457–1466. [Google Scholar] [CrossRef]
- Kim, S.; Park, M.; Yeom, S.I.; Kim, Y.M.; Lee, J.M.; Lee, H.A.; Seo, E.; Choi, J.; Cheong, K.; Kim, K.T.; et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 2014, 46, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Yu, C.; Shen, Y.; Fang, X.; Chen, L.; Min, J.; Cheng, J.; Zhao, S.; Xu, M.; Luo, Y.; et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc. Natl. Acad. Sci. USA 2014, 111, 5135–5140. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Nimmakayala, P.; Abburi, V.L.; Saminathan, T.; Alaparthi, S.B.; Almeida, A.; Davenport, B.; Nadimi, M.; Davidson, J.; Tonapi, K.; Yadav, V.; et al. Genome-wide diversity and association mapping for capsaicinoids and fruit weight in Capsicum annuum L. Sci. Rep. 2016, 6, 38081. [Google Scholar] [CrossRef] [PubMed]
- McLeod, L.; Barchi, L.; Tumino, G.; Tripodi, P.; Salinier, J.; Gros, C.; Boyaci, Y.C.; Ozalp, R.; Borovsky, Y.; Schafleitner, R.; et al. Multi-environment association study highlights candidate genes for robust agronomic quantitative trait loci in a novel worldwide Capsicum core collection. Plant J. 2023, 116, 1508–1528. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, B.; Huang, R.; Suo, H.; Zhang, Z.; Chen, W.; Dai, X.; Zou, X.; Ou, L. Transcriptome- and proteome-wide association of a recombinant inbred line population revealed twelve core QTLs for four fruit traits in pepper (Capsicum annuum L.). Hortic. Res. 2022, 9, uhac015. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ma, Q.; Mao, L.; Wu, Z.; Liu, Z.; Zou, X.; Yang, B. Comparative Transcriptome Analysis Identified Genes Associated with Fruit Size in Pepper (Capsicum annuum L.). Horticulturae 2023, 9, 1009. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.B.; Liu, K.W.; Diao, W.P.; Zhi, L.; Ge, W.; Liu, J.B.; Pan, B.G.; Wan, H.J.; Chen, J.F. Evaluation of appropriate reference genes for gene expression studies in pepper by quantitative real-time PCR. Mol. Breed. 2012, 30, 1393–1400. [Google Scholar]
- Brand, A.; Borovsky, Y.; Meir, S.; Rogachev, I.; Aharoni, A.; Paran, I. pc8.1, a major QTL for pigment content in pepper fruit, is associated with variation in plastid compartment size. Planta 2012, 235, 579–588. [Google Scholar] [CrossRef]
- Ma, X.; Qiao, Y.M.; Li, Y.; Yu, Y.N.; Gong, Z.H. Identification of Fruit Traits Related QTLs and a Candidate Gene, CaBRX, Controlling Locule Number in Pepper (Capsicum annuum L.). Horticulturae 2022, 8, 146. [Google Scholar] [CrossRef]
- Pereira, L.; Zhang, L.; Sapkota, M.; Ramos, A.; Razifard, H.; Caicedo, A.L.; van Der Knaap, E. Unraveling the genetics of tomato fruit weight during crop domestication and diversification. Theor. Appl. Genet. 2021, 134, 3363–3378. [Google Scholar] [CrossRef]
- Han, K.; Jeong, H.J.; Yang, H.B.; Kang, S.M.; Kwon, J.K.; Kim, S.; Choi, D.; Kang, B.C. An ultra-high-density bin map facilitates high-throughput QTL mapping of horticultural traits in pepper (Capsicum annuum). DNA Res. 2016, 23, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Hosmani, P.S.; Flores-Gonzalez, M.; van de Geest, H.; Maumus, F.; Bakker, L.V.; Schijlen, E.; van Haarst, J.; Cordewener, J.; Sanchez-Perez, G.; Peters, S.; et al. An improved de novo assembly and annotation of the tomato reference genome using single-molecule sequencing, Hi-C proximity ligation and optical maps. bioRxiv 2019, 767764. [Google Scholar] [CrossRef]
- Kim, J.H.; Tsukaya, H. Regulation of plant growth and development by the GROWTH-REGULATING FACTOR and GRF-INTERACTING FACTOR duo. J. Exp. Bot. 2015, 66, 6093–6107. [Google Scholar] [CrossRef]
- Van der Knaap, E.; Kim, J.H.; Kende, H. A Novel Gibberellin-Induced Gene from Rice and Its Potential Regulatory Role in Stem Growth. Plant Physiol. 2000, 122, 695–704. [Google Scholar] [CrossRef]
- Zhang, D.-F.; Li, B.; Jia, G.-Q.; Zhang, T.-F.; Dai, J.-R.; Li, J.-S.; Wang, S.-C. Isolation and characterization of genes encoding GRF transcription factors and GIF transcriptional coactivators in Maize (Zea mays L.). Plant Sci. 2008, 175, 809–817. [Google Scholar] [CrossRef]
- Aoi, Y.; Tanaka, K.; Cook, S.D.; Hayashi, K.I.; Kasahara, H. GH3 auxin-amido synthetases alter the ratio of indole-3-acetic acid and phenylacetic acid in Arabidopsis. Plant Cell Physiol. 2020, 61, 596–605. [Google Scholar] [CrossRef]
- Hua, B.; Wu, J.; Han, X.; Bian, X.; Xu, Z.; Sun, C.; Wang, R.; Zhang, W.; Liang, F.; Zhang, H.; et al. Auxin homeostasis is maintained by sly-miR167-SlARF8A/B-SlGH3. 4 feedback module in the development of locular and placental tissues of tomato fruits. New Phytol. 2024, 241, 1177–1192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Brewer, M.T.; van der Knaap, E. Fine mapping of fw3.2 controlling fruit weight in tomato. Theor. Appl. Genet. 2012, 125, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Adamski, N.M.; Anastasiou, E.; Eriksson, S.; O’Neill, C.M.; Lenhard, M. Local maternal control of seed size by KLUH/CYP78A5-dependent growth signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 20115–20120. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Population | Chr | LOD | PVE (%) | Left | Right |
|---|---|---|---|---|---|---|
| fruit weight without stalk | 2021SY47 | 2 | 10.60 | 12.40 | PepC2-152-3 | PepC2-156-1 |
| 2022SG136 | 14.43 | 14.61 | PepC2-150-2 | PepC2-154-2 | ||
| 2021SY47 | 3 | 12.87 | 14.97 | PepC3-12-1 | PepC3-18-1 | |
| 2022SG136 | 15.37 | 15.25 | PepC3-10-2 | PepC3-16-1 |
| Gene ID | Functional Prediction |
|---|---|
| Capana02g002936 | AP2-like ethylene-responsive transcription factor ANT |
| Capana02g002938 | growth-regulating factor 2-like (GRF) |
| Capana02g002971 | adenylyl-sulfate kinase |
| Capana02g002977 | mitochondrial outer membrane protein porin 2 |
| Capana02g002983 | domain-containing protein NPY2 |
| Capana02g002984 | GCN5-related N-acetyltransferase (GNAT) family protein |
| Capana02g002985 | U553_DICDI UPF0553 protein OS |
| Capana02g002986 | FMDA_METME Formamidase |
| Capana02g002991 | unknown protein |
| Capana02g003021 | probable indole-3-acetic acid-amido synthetase GH3.1 |
| Capana03g000827 | unknown protein |
| Capana03g000845 | NDH-DEPENDENT CYCLIC ELECTRON FLOW 1 |
| Capana03g000846 | UDP-glucose 6-dehydrogenase |
| Capana03g000898 | NLI interacting factor (NIF) family protein |
| Capana03g000903 | C78A4_PINRA Cytochrome P450 |
| Capana03g000937 | zinc finger (C2H2 type) family protein |
| Capana03g000971 | phosphoglycerate/bisphosphoglycerate mutase family protein |
| Capana03g000998 | ARATH Protein PRD1 |
| Capana03g0001010 | gibberellin-regulated protein 14 |
| Capana03g0001013 | calmodulin-binding protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, C.; Jin, Y.; Zhang, Z.; Cao, Y.; Wu, H.; Zhou, D.; Shao, W.; Yang, C.; Ban, G.; Ma, L.; et al. Fine Mapping and Candidate Gene Analysis of Two Major Quantitative Trait Loci, qFW2.1 and qFW3.1, Controlling Fruit Weight in Pepper (Capsicum annuum). Genes 2024, 15, 1097. https://doi.org/10.3390/genes15081097
Guan C, Jin Y, Zhang Z, Cao Y, Wu H, Zhou D, Shao W, Yang C, Ban G, Ma L, et al. Fine Mapping and Candidate Gene Analysis of Two Major Quantitative Trait Loci, qFW2.1 and qFW3.1, Controlling Fruit Weight in Pepper (Capsicum annuum). Genes. 2024; 15(8):1097. https://doi.org/10.3390/genes15081097
Chicago/Turabian StyleGuan, Congcong, Yuan Jin, Zhenghai Zhang, Yacong Cao, Huamao Wu, Daiyuan Zhou, Wenqi Shao, Chuangchuang Yang, Guoliang Ban, Lingling Ma, and et al. 2024. "Fine Mapping and Candidate Gene Analysis of Two Major Quantitative Trait Loci, qFW2.1 and qFW3.1, Controlling Fruit Weight in Pepper (Capsicum annuum)" Genes 15, no. 8: 1097. https://doi.org/10.3390/genes15081097
APA StyleGuan, C., Jin, Y., Zhang, Z., Cao, Y., Wu, H., Zhou, D., Shao, W., Yang, C., Ban, G., Ma, L., Wen, X., Chen, L., Cheng, S., Deng, Q., Yu, H., & Wang, L. (2024). Fine Mapping and Candidate Gene Analysis of Two Major Quantitative Trait Loci, qFW2.1 and qFW3.1, Controlling Fruit Weight in Pepper (Capsicum annuum). Genes, 15(8), 1097. https://doi.org/10.3390/genes15081097