


Bioinformatics for Inosine: Tools and Approaches to Trace This Elusive RNA Modification


Abstract
:1. Inosine and ADAR Protein Discovery

2. The Development of Inosine Detection Methods
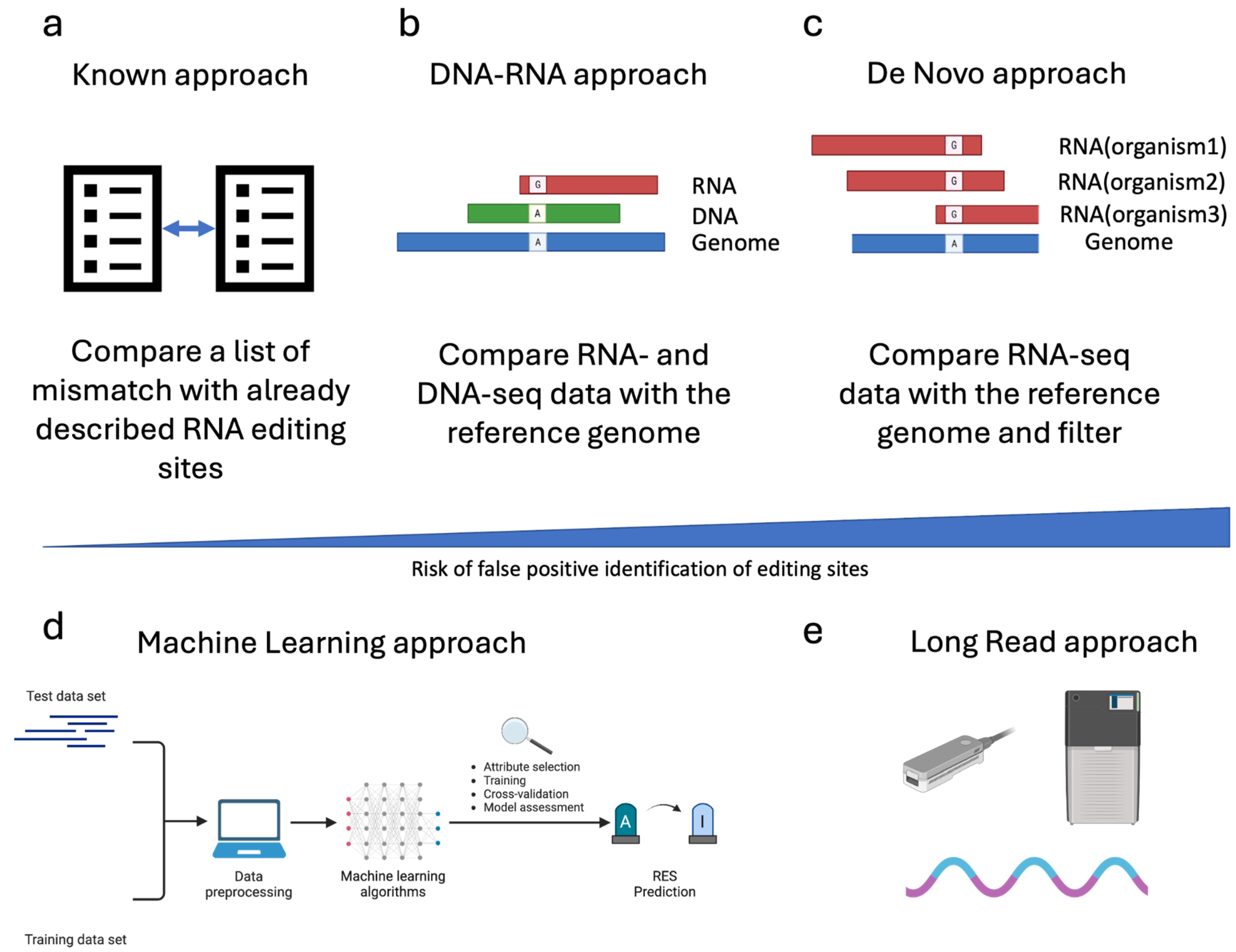
3. Strategies for Genome-Wide Identification of ADAR-Mediated RNA Editing Sites
3.1. File Preprocessing
3.2. Detection of RNA Editing Sites
3.3. RNA Editing Sites Filtering
4. RNA Editing Indexes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cohn, W.E. Pseudouridine, a Carbon-Carbon Linked Ribonucleoside in Ribonucleic Acids: Isolation, Structure, and Chemical Characteristics. J. Biol. Chem. 1960, 235, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Ontiveros, R.J.; Stoute, J.; Fange Liu, K. The Chemical Diversity of RNA Modifications. Biochem. J. 2019, 476, 1227–1245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, L.; Li, X. Detection Technologies for RNA Modifications. Exp. Mol. Med. 2022, 54, 1601–1616. [Google Scholar] [CrossRef] [PubMed]
- Boo, S.H.; Kim, Y.K. The Emerging Role of RNA Modifications in the Regulation of mRNA Stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef]
- Srinivas, K.P.; Depledge, D.P.; Abebe, J.S.; Rice, S.A.; Mohr, I.; Wilson, A.C. Widespread Remodeling of the m6A RNA-Modification Landscape by a Viral Regulator of RNA Processing and Export. Proc. Natl. Acad. Sci. USA 2021, 118, e2104805118. [Google Scholar] [CrossRef]
- Cui, L.; Ma, R.; Cai, J.; Guo, C.; Chen, Z.; Yao, L.; Wang, Y.; Fan, R.; Wang, X.; Shi, Y. RNA Modifications: Importance in Immune Cell Biology and Related Diseases. Signal Transduct. Target. Ther. 2022, 7, 334. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.; Emeson, R.B. RNA Editing. Annu. Rev. Neurosci. 1996, 19, 27–52. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.; Kapoor, U.; Jantsch, M.F. Understanding RNA Modifications: The Promises and Technological Bottlenecks of the ‘Epitranscriptome’. Open Biol. 2017, 7, 170077. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. A-to-I RNA Editing—Immune Protector and Transcriptome Diversifier. Nat. Rev. Genet. 2018, 19, 473–490. [Google Scholar] [CrossRef]
- Hartner, J.C.; Walkley, C.R.; Lu, J.; Orkin, S.H. ADAR1 Is Essential for the Maintenance of Hematopoiesis and Suppression of Interferon Signaling. Nat. Immunol. 2009, 10, 109–115. [Google Scholar] [CrossRef]
- Hwang, T.; Park, C.-K.; Leung, A.K.L.; Gao, Y.; Hyde, T.M.; Kleinman, J.E.; Rajpurohit, A.; Tao, R.; Shin, J.H.; Weinberger, D.R. Dynamic Regulation of RNA Editing in Human Brain Development and Disease. Nat. Neurosci. 2016, 19, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.J.; Shi, H.; He, C. Epitranscriptomic Influences on Development and Disease. Genome Biol. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Holley, R.W.; Apgar, J.; Everett, G.A.; Madison, J.T.; Marquisee, M.; Merrill, S.H.; Penswick, J.R.; Zamir, A. Structure of a Ribonucleic Acid. Science 1965, 147, 1462–1465. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.N.; Broni, E.; Miller, W.A. ADAR Family Proteins: A Structural Review. Curr. Issues Mol. Biol. 2024, 46, 3919–3945. [Google Scholar] [CrossRef]
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The Birth of the Epitranscriptome: Deciphering the Function of RNA Modifications. Genome Biol. 2012, 13, 175. [Google Scholar] [CrossRef]
- Bass, B. A Developmentally Regulated Activity That Unwinds RNA Duplexes. Cell 1987, 48, 607–613. [Google Scholar] [CrossRef]
- Rebagliati, M.R.; Melton, D.A. Antisense RNA Injections in Fertilized Frog Eggs Reveal an RNA Duplex Unwinding Activity. Cell 1987, 48, 599–605. [Google Scholar] [CrossRef]
- Bass, B.L.; Weintraub, H. An Unwinding Activity That Covalently Modifies Its Double-Stranded RNA Substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef]
- Hartner, J.C.; Schmittwolf, C.; Kispert, A.; Müller, A.M.; Higuchi, M.; Seeburg, P.H. Liver Disintegration in the Mouse Embryo Caused by Deficiency in the RNA-Editing Enzyme ADAR1. J. Biol. Chem. 2004, 279, 4894–4902. [Google Scholar] [CrossRef]
- Sommer, B.; Köhler, M.; Sprengel, R.; Seeburg, P.H. RNA Editing in Brain Controls a Determinant of Ion Flow in Glutamate-Gated Channels. Cell 1991, 67, 11–19. [Google Scholar] [CrossRef]
- Levanon, E.Y.; Eisenberg, E. Does RNA Editing Compensate for Alu Invasion of the Primate Genome? BioEssays 2015, 37, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Morse, D.P.; Bass, B.L. Detection of Inosine in Messenger RNA by Inosine-Specific Cleavage. Biochemistry 1997, 36, 8429–8434. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, G.; Lin, W.; Piskol, R.; Tan, M.H.; Davis, C.; Li, J.B. Accurate Identification of Human Alu and Non-Alu RNA Editing Sites. Nat. Methods 2012, 9, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, G.; Li, J.B. RADAR: A Rigorously Annotated Database of A-to-I RNA Editing. Nucleic Acids Res. 2014, 42, D109–D113. [Google Scholar] [CrossRef] [PubMed]
- Porath, H.T.; Carmi, S.; Levanon, E.Y. A Genome-Wide Map of Hyper-Edited RNA Reveals Numerous New Sites. Nat. Commun. 2014, 5, 4726. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Hur, B.; Kim, S. RDDpred: A Condition-Specific RNA-Editing Prediction Model from RNA-Seq Data. BMC Genom. 2016, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. Functions and Regulation of RNA Editing by ADAR Deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Wang, Y.; Chung, D.H.; Monteleone, L.R.; Li, J.; Chiang, Y.; Toney, M.D.; Beal, P.A. RNA Binding Candidates for Human ADAR3 from Substrates of a Gain of Function Mutant Expressed in Neuronal Cells. Nucleic Acids Res. 2019, 47, 10801–10814. [Google Scholar] [CrossRef]
- Picardi, E.; Pesole, G. REDItools: High-Throughput RNA Editing Detection Made Easy. Bioinformatics 2013, 29, 1813–1814. [Google Scholar] [CrossRef] [PubMed]
- Porath, H.T.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Massive A-to-I RNA Editing Is Common across the Metazoa and Correlates with dsRNA Abundance. Genome Biol. 2017, 18, 185. [Google Scholar] [CrossRef] [PubMed]
- Suspène, R.; Renard, M.; Henry, M.; Guétard, D.; Puyraimond-Zemmour, D.; Billecocq, A.; Bouloy, M.; Tangy, F.; Vartanian, J.-P.; Wain-Hobson, S. Inversing the Natural Hydrogen Bonding Rule to Selectively Amplify GC-Rich ADAR-Edited RNAs. Nucleic Acids Res. 2008, 36, e72. [Google Scholar] [CrossRef] [PubMed]
- Licht, K.; Hartl, M.; Amman, F.; Anrather, D.; Janisiw, M.P.; Jantsch, M.F. Inosine Induces Context-Dependent Recoding and Translational Stalling. Nucleic Acids Res. 2019, 47, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point Mutation in an AMPA Receptor Gene Rescues Lethality in Mice De®cient in the RNA-Editing Enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Li, I.-C.; Chen, Y.-C.; Wang, Y.-Y.; Tzeng, B.-W.; Ou, C.-W.; Lau, Y.-Y.; Wu, K.-M.; Chan, T.-M.; Lin, W.-H.; Hwang, S.-P.L.; et al. Zebrafish Adar2 Edits the Q/R Site of AMPA Receptor Subunit Gria2α Transcript to Ensure Normal Development of Nervous System and Cranial Neural Crest Cells. PLoS ONE 2014, 9, e97133. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, M.; Carlson, J.W.; Celniker, S.E. RNA Editing in Drosophila melanogaster: New Targets and Functional Consequences. RNA 2006, 12, 1922–1932. [Google Scholar] [CrossRef]
- Alon, S.; Garrett, S.C.; Levanon, E.Y.; Olson, S.; Graveley, B.R.; Rosenthal, J.J.C.; Eisenberg, E. The Majority of Transcripts in the Squid Nervous System Are Extensively Recoded by A-to-I RNA Editing. eLife 2015, 4, e05198. [Google Scholar] [CrossRef]
- Rangan, K.J.; Reck-Peterson, S.L. RNA Recoding in Cephalopods Tailors Microtubule Motor Protein Function. Cell 2023, 186, 2531–2543. [Google Scholar] [CrossRef]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA Editing of Alu-Containing mRNAs in the Human Transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef]
- Neeman, Y.; Levanon, E.Y.; Jantsch, M.F.; Eisenberg, E. RNA Editing Level in the Mouse Is Determined by the Genomic Repeat Repertoire. RNA 2006, 12, 1802–1809. [Google Scholar] [CrossRef]
- Bass, B.L. Adenosine Deaminases That Act on RNA, Then and Now. RNA 2024, 30, 521–529. [Google Scholar] [CrossRef]
- Baquero-Pérez, B.; Bortoletto, E.; Rosani, U.; Delgado-Tejedor, A.; Medina, R.; Novoa, E.M.; Venier, P.; Díez, J. Elucidation of the Epitranscriptomic RNA Modification Landscape of Chikungunya Virus. Viruses 2024, 16, 945. [Google Scholar] [CrossRef]
- Min, Y.-H.; Shao, W.-X.; Hu, Q.-S.; Xie, N.-B.; Zhang, S.; Feng, Y.-Q.; Xing, X.-W.; Yuan, B.-F. Simultaneous Detection of Adenosine-to-Inosine Editing and N6-Methyladenosine at Identical RNA Sites through Deamination-Assisted Reverse Transcription Stalling. Anal. Chem. 2024, 96, 8730–8739. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Suzuki, T. Biochemical Identification of A-to-I RNA Editing Sites by the Inosine Chemical Erasing (ICE) Method. In RNA and DNA Editing: Methods and Protocols; Aphasizhev, R., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 89–99. ISBN 978-1-61779-018-8. [Google Scholar]
- Ding, J.-H.; Chen, M.-Y.; Xie, N.-B.; Xie, C.; Xiong, N.; He, J.-G.; Wang, J.; Guo, C.; Feng, Y.-Q.; Yuan, B.-F. Quantitative and Site-Specific Detection of Inosine Modification in RNA by Acrylonitrile Labeling-Mediated Elongation Stalling. Biosens. Bioelectron. 2023, 219, 114821. [Google Scholar] [CrossRef]
- Chen, J.-J.; You, X.-J.; Li, L.; Xie, N.-B.; Ding, J.-H.; Yuan, B.-F.; Feng, Y.-Q. Single-Base Resolution Detection of Adenosine-to-Inosine RNA Editing by Endonuclease-Mediated Sequencing. Anal. Chem. 2022, 94, 8740–8747. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality Control of RNA-Seq Experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef]
- DeLuca, D.S.; Levin, J.Z.; Sivachenko, A.; Fennell, T.; Nazaire, M.-D.; Williams, C.; Reich, M.; Winckler, W.; Getz, G. RNA-SeQC: RNA-Seq Metrics for Quality Control and Process Optimization. Bioinformatics 2012, 28, 1530–1532. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A Fast and Accurate Adapter Trimmer for next-Generation Sequencing Paired-End Reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Sahlin, K.; Baudeau, T.; Cazaux, B.; Marchet, C. A Survey of Mapping Algorithms in the Long-Reads Era. Genome Biol. 2023, 24, 133. [Google Scholar] [CrossRef]
- Lo Giudice, C.; Silvestris, D.A.; Roth, S.H.; Eisenberg, E.; Pesole, G.; Gallo, A.; Picardi, E. Quantifying RNA Editing in Deep Transcriptome Datasets. Front. Genet. 2020, 11, 194. [Google Scholar] [CrossRef]
- Lo Giudice, C.; Tangaro, M.A.; Pesole, G.; Picardi, E. Investigating RNA Editing in Deep Transcriptome Datasets with REDItools and REDIportal. Nat. Protoc. 2020, 15, 1098–1131. [Google Scholar] [CrossRef]
- Picardi, E.; Pesole, G. (Eds.) RNA Editing: Methods and Protocols; Methods in Molecular Biology; Springer US: New York, NY, USA, 2021; Volume 2181, ISBN 978-1-07-160786-2. [Google Scholar]
- Morales, D.R.; Rennie, S.; Uchida, S. Benchmarking RNA Editing Detection Tools. BioTech 2023, 12, 56. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinforma. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Picardi, E.; D’Erchia, A.M.; Lo Giudice, C.; Pesole, G. REDIportal: A Comprehensive Database of A-to-I RNA Editing Events in Humans. Nucleic Acids Res. 2017, 45, D750–D757. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lian, J.; Li, Q.; Zhang, P.; Zhou, Y.; Zhan, X.; Zhang, G. RES-Scanner: A Software Package for Genome-Wide Identification of RNA-Editing Sites. GigaScience 2016, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Piechotta, M.; Wyler, E.; Ohler, U.; Landthaler, M.; Dieterich, C. JACUSA: Site-Specific Identification of RNA Editing Events from Replicate Sequencing Data. BMC Bioinform. 2017, 18, 7. [Google Scholar] [CrossRef]
- Light, D.; Haas, R.; Yazbak, M.; Elfand, T.; Blau, T.; Lamm, A.T. RESIC: A Tool for Comprehensive Adenosine to Inosine RNA Editing Site Identification and Classification. Front. Genet. 2021, 12, 686851. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xiao, X. Genome Sequence–Independent Identification of RNA Editing Sites. Nat. Methods 2015, 12, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Lu, Y.; Yan, S.; Xing, Q.; Tian, W. SPRINT: An SNP-Free Toolkit for Identifying RNA Editing Sites. Bioinformatics 2017, 33, 3538–3548. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Liu, D.; Li, Q.; Lei, M.; Xu, L.; Wu, L.; Wang, Z.; Ren, S.; Li, W.; Xia, M.; et al. RED-ML: A Novel, Effective RNA Editing Detection Method Based on Machine Learning. GigaScience 2017, 6, gix012. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Liu, F.; Zhao, C.; Ren, C.; An, G.; Mei, C.; Bo, X.; Shu, W. Accurate Identification of RNA Editing Sites from Primitive Sequence with Deep Neural Networks. Sci. Rep. 2018, 8, 6005. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Heng, J.W.J.; Kaewsapsak, P.; Kok, E.P.L.; Stanojević, D.; Liu, H.; Cardilla, A.; Praditya, A.; Yi, Z.; Lin, M.; et al. Direct Identification of A-to-I Editing Sites with Nanopore Native RNA Sequencing. Nat. Methods 2022, 19, 833–844. [Google Scholar] [CrossRef]
- Chen, L.; Ou, L.; Jing, X.; Kong, Y.; Xie, B.; Zhang, N.; Shi, H.; Qin, H.; Li, X.; Hao, P. DeepEdit: Single-Molecule Detection and Phasing of A-to-I RNA Editing Events Using Nanopore Direct RNA Sequencing. Genome Biol. 2023, 24, 75. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Quinones-Valdez, G.; Fu, T.; Huang, E.; Choudhury, M.; Reese, F.; Mortazavi, A.; Xiao, X. L-GIREMI Uncovers RNA Editing Sites in Long-Read RNA-Seq. Genome Biol. 2023, 24, 171. [Google Scholar] [CrossRef] [PubMed]
- Buchumenski, I.; Holler, K.; Appelbaum, L.; Eisenberg, E.; Junker, J.P.; Levanon, E.Y. Systematic Identification of A-to-I RNA Editing in Zebrafish Development and Adult Organs. Nucleic Acids Res. 2021, 49, 4325–4337. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Heng, J.W.J.; Ng, Y.T.; Sun, R.; Fisher, S.; Oguz, G.; Kaewsapsak, P.; Xue, S.; Reversade, B.; Ramasamy, A.; et al. Deep Transcriptome Profiling Reveals Limited Conservation of A-to-I RNA Editing in Xenopus. BMC Biol. 2023, 21, 251. [Google Scholar] [CrossRef] [PubMed]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic Identification of Abundant A-to-I Editing Sites in the Human Transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.H.; Levanon, E.Y.; Eisenberg, E. Genome-Wide Quantification of ADAR Adenosine-to-Inosine RNA Editing Activity. Nat. Methods 2019, 16, 1131–1138. [Google Scholar] [CrossRef]
- Silvestris, D.A.; Picardi, E.; Cesarini, V.; Fosso, B.; Mangraviti, N.; Massimi, L.; Martini, M.; Pesole, G.; Locatelli, F.; Gallo, A. Dynamic Inosinome Profiles Reveal Novel Patient Stratification and Gender-Specific Differences in Glioblastoma. Genome Biol. 2019, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Ding, H.; Lin, H.; Chou, K.-C. iRNA-Methyl: Identifying N(6)-Methyladenosine Sites Using Pseudo Nucleotide Composition. Anal. Biochem. 2015, 490, 26–33. [Google Scholar] [CrossRef]
- Chen, W.; Tran, H.; Liang, Z.; Lin, H.; Zhang, L. Identification and Analysis of the N(6)-Methyladenosine in the Saccharomyces Cerevisiae Transcriptome. Sci. Rep. 2015, 5, 13859. [Google Scholar] [CrossRef]
- Li, Y.-H.; Zhang, G.; Cui, Q. PPUS: A Web Server to Predict PUS-Specific Pseudouridine Sites. Bioinforma. Oxf. Engl. 2015, 31, 3362–3364. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Tang, H.; Ding, H.; Lin, H. RAMPred: Identifying the N(1)-Methyladenosine Sites in Eukaryotic Transcriptomes. Sci. Rep. 2016, 6, 31080. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Ding, H.; Lin, H. Identifying N 6-Methyladenosine Sites in the Arabidopsis Thaliana Transcriptome. Mol. Genet. Genom. MGG 2016, 291, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.-Z.; Zhang, J.-J.; Gu, W.-Z. RNA-MethylPred: A High-Accuracy Predictor to Identify N6-Methyladenosine in RNA. Anal. Biochem. 2016, 510, 72–75. [Google Scholar] [CrossRef]
- Li, G.-Q.; Liu, Z.; Shen, H.-B.; Yu, D.-J. TargetM6A: Identifying N6-Methyladenosine Sites From RNA Sequences via Position-Specific Nucleotide Propensities and a Support Vector Machine. IEEE Trans. Nanobiosci. 2016, 15, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiao, X.; Yu, D.-J.; Jia, J.; Qiu, W.-R.; Chou, K.-C. pRNAm-PC: Predicting N(6)-Methyladenosine Sites in RNA Sequences via Physical-Chemical Properties. Anal. Biochem. 2016, 497, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Liu, K.; Yan, Z.; Zhang, Y.; Sun, Z. RNAMethPre: A Web Server for the Prediction and Query of mRNA m6A Sites. PLoS ONE 2016, 11, e0162707. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Yan, Z.; Liu, K.; Zhang, Y.; Sun, Z. AthMethPre: A Web Server for the Prediction and Query of mRNA m6A Sites in Arabidopsis Thaliana. Mol. Biosyst. 2016, 12, 3333–3337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, J.-W.; Liu, Z.; Ren, M.-W.; Shen, H.-B.; Yu, D.-J. Improving N(6)-Methyladenosine Site Prediction with Heuristic Selection of Nucleotide Physical-Chemical Properties. Anal. Biochem. 2016, 508, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zeng, P.; Li, Y.-H.; Zhang, Z.; Cui, Q. SRAMP: Prediction of Mammalian N6-Methyladenosine (m6A) Sites Based on Sequence-Derived Features. Nucleic Acids Res. 2016, 44, e91. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Tang, H.; Ding, H.; Lin, H. Identifying 2’-O-Methylationation Sites by Integrating Nucleotide Chemical Properties and Nucleotide Compositions. Genomics 2016, 107, 255–258. [Google Scholar] [CrossRef]
- Chen, W.; Tang, H.; Ye, J.; Lin, H.; Chou, K.-C. iRNA-PseU: Identifying RNA Pseudouridine Sites. Mol. Ther. Nucleic Acids 2016, 5, e332. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.-R.; Jiang, S.-Y.; Xu, Z.-C.; Xiao, X.; Chou, K.-C. iRNAm5C-PseDNC: Identifying RNA 5-Methylcytosine Sites by Incorporating Physical-Chemical Properties into Pseudo Dinucleotide Composition. Oncotarget 2017, 8, 41178–41188. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Ding, H.; Yang, H.; Chen, W.; Lin, H.; Chou, K.-C. iRNA-PseColl: Identifying the Occurrence Sites of Different RNA Modifications by Incorporating Collective Effects of Nucleotides into PseKNC. Mol. Ther. Nucleic Acids 2017, 7, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, H.; Lin, H. MethyRNA: A Web Server for Identification of N6-Methyladenosine Sites. J. Biomol. Struct. Dyn. 2017, 35, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xing, P.; Zou, Q. Detecting N6-Methyladenosine Sites from RNA Transcriptomes Using Ensemble Support Vector Machines. Sci. Rep. 2017, 7, 40242. [Google Scholar] [CrossRef] [PubMed]
- Xing, P.; Su, R.; Guo, F.; Wei, L. Identifying N6-Methyladenosine Sites Using Multi-Interval Nucleotide Pair Position Specificity and Support Vector Machine. Sci. Rep. 2017, 7, 46757. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chou, K.-C. iRNA-3typeA: Identifying Three Types of Modification at RNA’s Adenosine Sites. Mol. Ther. Nucleic Acids 2018, 11, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhai, J.; Bian, E.; Song, Y.; Yu, J.; Ma, C. Transcriptome-Wide Annotation of m5C RNA Modifications Using Machine Learning. Front. Plant Sci. 2018, 9, 519. [Google Scholar] [CrossRef]
- Sabooh, M.F.; Iqbal, N.; Khan, M.; Khan, M.; Maqbool, H.F. Identifying 5-Methylcytosine Sites in RNA Sequence Using Composite Encoding Feature into Chou’s PseKNC. J. Theor. Biol. 2018, 452, 1–9. [Google Scholar] [CrossRef]
- Li, J.; Huang, Y.; Yang, X.; Zhou, Y.; Zhou, Y. RNAm5Cfinder: A Web-Server for Predicting RNA 5-Methylcytosine (m5C) Sites Based on Random Forest. Sci. Rep. 2018, 8, 17299. [Google Scholar] [CrossRef]
- Akbar, S.; Hayat, M. iMethyl-STTNC: Identification of N6-Methyladenosine Sites by Extending the Idea of SAAC into Chou’s PseAAC to Formulate RNA Sequences. J. Theor. Biol. 2018, 455, 205–211. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Zhou, X.; Lin, H.; Chou, K.-C. iRNA(m6A)-PseDNC: Identifying N6-Methyladenosine Sites Using Pseudo Dinucleotide Composition. Anal. Biochem. 2018, 561–562, 59–65. [Google Scholar] [CrossRef]
- Zhao, Z.; Peng, H.; Lan, C.; Zheng, Y.; Fang, L.; Li, J. Imbalance Learning for the Prediction of N6-Methylation Sites in mRNAs. BMC Genomics 2018, 19, 574. [Google Scholar] [CrossRef]
- Wei, L.; Chen, H.; Su, R. M6APred-EL: A Sequence-Based Predictor for Identifying N6-Methyladenosine Sites Using Ensemble Learning. Mol. Ther. Nucleic Acids 2018, 12, 635–644. [Google Scholar] [CrossRef]
- Huang, Y.; He, N.; Chen, Y.; Chen, Z.; Li, L. BERMP: A Cross-Species Classifier for Predicting m6A Sites by Integrating a Deep Learning Algorithm and a Random Forest Approach. Int. J. Biol. Sci. 2018, 14, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.; Chen, H.; Ye, X.; Su, R.; Wei, L. M6AMRFS: Robust Prediction of N6-Methyladenosine Sites With Sequence-Based Features in Multiple Species. Front. Genet. 2018, 9, 495. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yan, R. RFAthM6A: A New Tool for Predicting m6A Sites in Arabidopsis Thaliana. Plant Mol. Biol. 2018, 96, 327–337. [Google Scholar] [CrossRef]
- Yang, H.; Lv, H.; Ding, H.; Chen, W.; Lin, H. iRNA-2OM: A Sequence-Based Predictor for Identifying 2’-O-Methylation Sites in Homo Sapiens. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2018, 25, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cui, Q.; Zhou, Y. NmSEER: A Prediction Tool for 2’-O-Methylation (Nm) Sites Based on Random Forest. In Proceedings of the Intelligent Computing Theories and Application: 14th International Conference, ICIC 2018, Wuhan, China, 15–18 August 2018; Proceedings, Part I. Springer-Verlag: Berlin, Heidelberg, 2018; pp. 893–900. [Google Scholar]
- He, J.; Fang, T.; Zhang, Z.; Huang, B.; Zhu, X.; Xiong, Y. PseUI: Pseudouridine Sites Identification Based on RNA Sequence Information. BMC Bioinform. 2018, 19, 306. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Zhang, Z.; Sun, R.; Zhu, L.; He, J.; Huang, B.; Xiong, Y.; Zhu, X. RNAm5CPred: Prediction of RNA 5-Methylcytosine Sites Based on Three Different Kinds of Nucleotide Composition. Mol. Ther. Nucleic Acids 2019, 18, 739–747. [Google Scholar] [CrossRef]
- Sun, P.P.; Chen, Y.B.; Liu, B.; Gao, Y.X.; Han, Y.; He, F.; Ji, J.C. DeepMRMP: A New Predictor for Multiple Types of RNA Modification Sites Using Deep Learning. Math. Biosci. Eng. MBE 2019, 16, 6231–6241. [Google Scholar] [CrossRef]
- Wei, L.; Su, R.; Wang, B.; Li, X.; Zou, Q.; Gao, X. Integration of Deep Feature Representations and Handcrafted Features to Improve the Prediction of N6-Methyladenosine Sites. Neurocomputing 2019, 324, 3–9. [Google Scholar] [CrossRef]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene Subsequence Embedding for Prediction of Mammalian N6-Methyladenosine Sites from mRNA. RNA N. Y. N 2019, 25, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; Zhang, S.-W.; Fan, X.-N.; Zhang, T.; Meng, J.; Huang, Y. FunDMDeep-m6A: Identification and Prioritization of Functional Differential m6A Methylation Genes. Bioinformatics 2019, 35, i90–i98. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Song, X.; Lv, H.; Lin, H. iRNA-m7G: Identifying N7-Methylguanosine Sites by Fusing Multiple Features. Mol. Ther. Nucleic Acids 2019, 18, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.; Tayara, H.; Chong, K.T. iRNA-PseKNC(2methyl): Identify RNA 2’-O-Methylation Sites by Convolution Neural Network and Chou’s Pseudo Components. J. Theor. Biol. 2019, 465, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cui, Q.; Zhou, Y. NmSEER V2.0: A Prediction Tool for 2’-O-Methylation Sites Based on Random Forest and Multi-Encoding Combination. BMC Bioinform. 2019, 20, 690. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.; Tayara, H.; Chong, K.T. iPseU-CNN: Identifying RNA Pseudouridine Sites Using Convolutional Neural Networks. Mol. Ther. Nucleic Acids 2019, 16, 463–470. [Google Scholar] [CrossRef]
- Nguyen-Vo, T.-H.; Nguyen, Q.H.; Do, T.T.T.; Nguyen, T.-N.; Rahardja, S.; Nguyen, B.P. iPseU-NCP: Identifying RNA Pseudouridine Sites Using Random Forest and NCP-Encoded Features. BMC Genom. 2019, 20, 971. [Google Scholar] [CrossRef]
- Chen, K.; Wei, Z.; Zhang, Q.; Wu, X.; Rong, R.; Lu, Z.; Su, J.; de Magalhães, J.P.; Rigden, D.J.; Meng, J. WHISTLE: A High-Accuracy Map of the Human N6-Methyladenosine (m6A) Epitranscriptome Predicted Using a Machine Learning Approach. Nucleic Acids Res. 2019, 47, e41. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, Z.-M.; Li, S.-H.; Tan, J.-X.; Chen, W.; Lin, H. Evaluation of Different Computational Methods on 5-Methylcytosine Sites Identification. Brief. Bioinform. 2020, 21, 982–995. [Google Scholar] [CrossRef]
- Chen, X.; Xiong, Y.; Liu, Y.; Chen, Y.; Bi, S.; Zhu, X. m5CPred-SVM: A Novel Method for Predicting m5C Sites of RNA. BMC Bioinformatics 2020, 21, 489. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Song, B.; Tang, Y.; Chen, K.; Wei, Z.; Meng, J. m5UPred: A Web Server for the Prediction of RNA 5-Methyluridine Sites from Sequences. Mol. Ther. Nucleic Acids 2020, 22, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Li, F.; Wang, Y.; Smith, A.I.; Webb, G.I.; Akutsu, T.; Baggag, A.; Bensmail, H.; Song, J. Comprehensive Review and Assessment of Computational Methods for Predicting RNA Post-Transcriptional Modification Sites from RNA Sequences. Brief. Bioinform. 2020, 21, 1676–1696. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Chen, W. iMRM: A Platform for Simultaneously Identifying Multiple Kinds of RNA Modifications. Bioinforma. Oxf. Engl. 2020, 36, 3336–3342. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, Z.; Mao, X.; Li, Q. m7GPredictor: An Improved Machine Learning-Based Model for Predicting Internal m7G Modifications Using Sequence Properties. Anal. Biochem. 2020, 609, 113905. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Xiang, D.; Ge, Z.; Li, F.; Jia, C.; Song, J. An Interpretable Prediction Model for Identifying N7-Methylguanosine Sites Based on XGBoost and SHAP. Mol. Ther. Nucleic Acids 2020, 22, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Jin, D.; Jia, C. EnsemPseU: Identifying Pseudouridine Sites With an Ensemble Approach. IEEE Access 2020, 8, 79376–79382. [Google Scholar] [CrossRef]
- Song, B.; Tang, Y.; Wei, Z.; Liu, G.; Su, J.; Meng, J.; Chen, K. PIANO: A Web Server for Pseudouridine-Site (Ψ) Identification and Functional Annotation. Front. Genet. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Chen, K.; Tang, Y.; Ma, J.; Meng, J.; Wei, Z. PSI-MOUSE: Predicting Mouse Pseudouridine Sites From Sequence and Genome-Derived Features. Evol. Bioinforma. Online 2020, 16, 1176934320925752. [Google Scholar] [CrossRef]
- Liu, K.; Chen, W.; Lin, H. XG-PseU: An eXtreme Gradient Boosting Based Method for Identifying Pseudouridine Sites. Mol. Genet. Genomics MGG 2020, 295, 13–21. [Google Scholar] [CrossRef]
- Lv, Z.; Zhang, J.; Ding, H.; Zou, Q. RF-PseU: A Random Forest Predictor for RNA Pseudouridine Sites. Front. Bioeng. Biotechnol. 2020, 8, 134. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.U.; Hong, K.J.; Tayara, H.; Chong, K. to m6A-NeuralTool: Convolution Neural Tool for RNA N6-Methyladenosine Site Identification in Different Species. IEEE Access 2021, 9, 17779–17786. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, X.; Liu, M.; Xu, Z.; Liu, G. DNN-m6A: A Cross-Species Method for Identifying RNA N6-Methyladenosine Sites Based on Deep Neural Network with Multi-Information Fusion. Genes 2021, 12, 354. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Feng, P.; Cui, L.; Su, R.; Chen, W.; Wei, L. Iterative Feature Representation Algorithm to Improve the Predictive Performance of N7-Methylguanosine Sites. Brief. Bioinform. 2021, 22, bbaa278. [Google Scholar] [CrossRef] [PubMed]
- Ning, Q.; Sheng, M. m7G-DLSTM: Intergrating Directional Double-LSTM and Fully Connected Network for RNA N7-Methlguanosine Sites Prediction in Human. Chemom. Intell. Lab. Syst. 2021, 217, 104398. [Google Scholar] [CrossRef]
- Li, H.; Chen, L.; Huang, Z.; Luo, X.; Li, H.; Ren, J.; Xie, Y. DeepOMe: A Web Server for the Prediction of 2’-O-Me Sites Based on the Hybrid CNN and BLSTM Architecture. Front. Cell Dev. Biol. 2021, 9, 686894. [Google Scholar] [CrossRef] [PubMed]
- Begik, O.; Lucas, M.C.; Pryszcz, L.P.; Ramirez, J.M.; Medina, R.; Milenkovic, I.; Cruciani, S.; Liu, H.; Vieira, H.G.S.; Sas-Chen, A.; et al. Quantitative Profiling of Pseudouridylation Dynamics in Native RNAs with Nanopore Sequencing. Nat. Biotechnol. 2021, 39, 1278–1291. [Google Scholar] [CrossRef]
- Feng, P.; Chen, W. iRNA-m5U: A Sequence Based Predictor for Identifying 5-Methyluridine Modification Sites in Saccharomyces Cerevisiae. Methods San Diego Calif 2022, 203, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Rehman, H.U.; Habib, U.; Ijaz, U. m6A-Finder: Detecting m6A Methylation Sites from RNA Transcriptomes Using Physical and Statistical Properties Based Features. Comput. Biol. Chem. 2022, 97, 107640. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Basith, S.; Pitti, T.; Lee, G.; Manavalan, B. THRONE: A New Approach for Accurate Prediction of Human RNA N7-Methylguanosine Sites. J. Mol. Biol. 2022, 434, 167549. [Google Scholar] [CrossRef]
- Ao, C.; Zou, Q.; Yu, L. NmRF: Identification of Multispecies RNA 2’-O-Methylation Modification Sites from RNA Sequences. Brief. Bioinform. 2022, 23, bbab480. [Google Scholar] [CrossRef] [PubMed]
- Hassan, D.; Acevedo, D.; Daulatabad, S.V.; Mir, Q.; Janga, S.C. Penguin: A Tool for Predicting Pseudouridine Sites in Direct RNA Nanopore Sequencing Data. Methods 2022, 203, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Ao, C.; Ye, X.; Sakurai, T.; Zou, Q.; Yu, L. m5U-SVM: Identification of RNA 5-Methyluridine Modification Sites Based on Multi-View Features of Physicochemical Features and Distributed Representation. BMC Biol. 2023, 21, 93. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chung, C.-R.; Huang, H.-D.; Lee, T.-Y. Identification of Species-Specific RNA N6-Methyladinosine Modification Sites from RNA Sequences. Brief. Bioinform. 2023, 24, bbac573. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.U.; Tayara, H.; Chong, K.T. DL-m6A: Identification of N6-Methyladenosine Sites in Mammals Using Deep Learning Based on Different Encoding Schemes. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zhao, Y.; Jin, J.; Qiao, J.; Wang, D.; Wang, Y.; Wei, L. Rm-LR: A Long-Range-Based Deep Learning Model for Predicting Multiple Types of RNA Modifications. Comput. Biol. Med. 2023, 164, 107238. [Google Scholar] [CrossRef] [PubMed]
- Soylu, N.N.; Sefer, E. BERT2OME: Prediction of 2’-O-Methylation Modifications From RNA Sequence by Transformer Architecture Based on BERT. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 2177–2189. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Ma, C.-Y.; Gao, D.; Liu, X.-W.; Yuan, S.-S.; Ding, H. i2OM: Toward a Better Prediction of 2’-O-Methylation in Human RNA. Int. J. Biol. Macromol. 2023, 239, 124247. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.T.; Rakkiyapan, R.; Park, J.; Malik, A.; Manavalan, B. H2Opred: A Robust and Efficient Hybrid Deep Learning Model for Predicting 2’-O-Methylation Sites in Human RNA. Brief. Bioinform. 2023, 25, bbad476. [Google Scholar] [CrossRef]
- Tu, G.; Wang, X.; Xia, R.; Song, B. m6A-TCPred: A Web Server to Predict Tissue-Conserved Human m6A Sites Using Machine Learning Approach. BMC Bioinform. 2024, 25, 127. [Google Scholar] [CrossRef]
- Wang, H.; Huang, T.; Wang, D.; Zeng, W.; Sun, Y.; Zhang, L. MSCAN: Multi-Scale Self- and Cross-Attention Network for RNA Methylation Site Prediction. BMC Bioinform. 2024, 25, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xu, Y.; Liang, Y. TMSC-m7G: A Transformer Architecture Based on Multi-Sense-Scaled Embedding Features and Convolutional Neural Network to Identify RNA N7-Methylguanosine Sites. Comput. Struct. Biotechnol. J. 2024, 23, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jin, J.; Gao, W.; Qiao, J.; Wei, L. Moss-m7G: A Motif-Based Interpretable Deep Learning Method for RNA N7-Methlguanosine Site Prediction. J. Chem. Inf. Model. 2024. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA Modification | Tool |
|---|---|
| m1A | RAMPred, iRNA-3typeA, DeepPromise, MSCAN, Rm-LR, iMRM |
| m5C | iRNAm5C-PseDNC, iRNA- PseColl, PEA-m5C, pM5CS-Comp-mRMR, RNAm5Cfinder, RNAm5CPred, DeepMRMP, iRNA-m5C, m5CPred-SVM, MSCAN, Rm-LR, iMRM |
| m5U | iRNA-m5U, m5UPred, m5U-SVM, MSCAN, Rm-LR |
| m6A | iRNA-3typeA, iRNA-Methyl, m6Apred, M6ATH, RNA-MethylPred, TargetM6A, pRNAm-PC, RNAMethPre, AthMethPre, M6A-HPCS, SRAMP, MethyRNA, RAM-ESVM, RAM-NPPS, iMethyl-STTNC, iRNA(m6A)-PseDNC, HMpre, M6APred-EL, BERMP, M6AMRFS, RFAthM6A, DeepM6APred, Gene2Vec, FunDMDeep-m6A, m6A-NeuralTool, DNN-m6A, WHISTLE, Adaptive-m6A, DL-m6A, m6A-Finder, m6A-TCPred, DeepPromise, MSCAN, Rm-LR, iMRM |
| m6Am | MSCAN, Rm-LR |
| m7G | MSCAN, iRNA-m7G, m7GPredictor, XG-m7G, m7G-IFL, m7G-DLSTM, THRONE, TMSC-m7G, Moss-m7G |
| Ψ | MSCAN, iMRM, PPUS, iRNA-PseU, PseUI, iPseU-CNN, iPseU-NCP, EnsemPseU, PIANO, PSI-MOUSE, XG-PseU, RF-PseU, NanoPsu, Penguin |
| Nm | Rm-LR, iRNA-2OM, NmSEER, iRNA-PseKNC(2methyl), NmSEER V2.0, DeepOMe, NmRF, BERT2OME, i2OM, H2Opred |
| Method | Type | Description | Limitations |
|---|---|---|---|
| Thin-Layer Chromatography (2D-TLC) [42] | Direct | Detect RNA modifications by separating nucleotides based on their mobility in a solvent. RNA is partially digested into oligonucleotides, labelled with 32P and separated via 2D-TLC. Nucleotides are identified by comparing their retardation factors to standards and quantified by measuring radioactivity. | -Low throughput and labor-intensive. -Requires radioactive labeling, which involves safety and disposal issues. -Limited sensitivity and resolution compared to modern high-throughput methods. |
| Inosine-Specific Cleavage Assays [23] | Direct | This method involves treating RNA with chemicals or enzymes that specifically cleave at inosine sites. For instance, inosine-specific ribonucleases can be used to cut RNA at inosine residues, allowing for the identification of inosine locations. | -Limited to specific cleavage sites, potentially missing some inosine modifications. -Requires precise enzymatic or chemical conditions, which can be challenging to optimize. -Not suitable for high-throughput analysis. |
| Mass Spectrometry (MS) [43] | Direct | Identifies and quantifies inosine by analyzing the mass-to-charge ratio of RNA fragments. | -Can be complex and time-consuming to prepare samples and interpret results. -Sensitivity can be an issue for low-abundance modifications. |
| DARTS [44] | Direct | Allows the concurrent quantification of A-to-I editing and m6A modifications at the same sites in RNA. | -Limited to single-site analysis and cannot be used for transcriptome-wide analysis of RNA modifications. |
| ICE-seq [45] | Direct | Biochemical method useful for identifying inosines based on cyanoethylation combined with reverse transcription and RNA-seq. | -Requires chemical modification of RNA, which can be technically challenging. |
| ALES [46] | Direct | The ALES method detects RNA editing by modifying inosine to N1-cyanoethylinosine, which causes reverse transcription to stall at the edited site. This stalling is then measured using real-time quantitative PCR, allowing precise identification and quantification of RNA editing. | -ALES method is not capable of transcriptome-wide mapping of inosine sites. |
| hEndoV-seq [47] | Direct | This approach blocks the RNA terminal 3′OH using 3′-deoxyadenosine. The hEndoV protein then specifically cleaves inosine sites, creating new terminal 3′OH ends. These new ends can be identified through sequencing analysis, allowing site-specific inosine detection in RNA. | -Limited throughput, not ideal for large-scale studies. |
| Sanger Sequencing [48] | Indirect | Detects A-to-G mismatches after reverse transcribing RNA to cDNA. | -Low throughput and labor-intensive compared to next-generation sequencing -Limited sensitivity, especially for low-frequency editing events. |
| Next-Generation Sequencing (NGS) [24] | Indirect | High-throughput sequencing of cDNA. A-to-G transitions are then traced with a specific bioinformatic pipeline. | -Requires significant computational resources and bioinformatic expertise -Potential for sequencing errors and alignment ambiguities, leading to false positives. |
| Nanopore direct RNA sequencing | Direct | Direct sequencing of native RNA, inosine can be directly detected from the raw electric signals. | -Lack of an inosine-dedicated basecaller. |
| Tool Name | Input Datasets | Description |
|---|---|---|
| REDItools [31] | DNA-RNA RNA | REDItools provides a nice output table and a series of accessory scripts for annotating and filtering the resulting RES. |
| RES-Scanner [65] | DNA-RNA | RES-Scanner uses a combination of different statistical models for homozygous genotype calling and filters to remove potential false-positive RES, also providing an associated p-value. |
| JACUSA [66] | DNA-RNA RNA | JACUSA is designed with user-friendliness in mind. It offers a streamlined process for detecting RNA editing events by exploiting a robust statistical approach. |
| RESIC [67] | DNA-RNA RNA | RESIC allows the exclusion of polymorphism sites so as to increase reliability based on DNA-seq, ADAR-mutant RNA-seq datasets or SNP databases. |
| GIREMI [68] | RNA | GIREMI can integrate biological replicates into one dataset for higher accuracy in RNA editing detection. Its algorithm is based on mutual information between editing sites in RNA-seq data and SNPs and is thus suitable only for diploid genomes. |
| SPRINT [69] | RNA | SPRINT can identify RNA editing sites, without the need to utilize SNP databases, by clustering RES and SNP duplets based on their distinctive and unique distribution. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bortoletto, E.; Rosani, U. Bioinformatics for Inosine: Tools and Approaches to Trace This Elusive RNA Modification. Genes 2024, 15, 996. https://doi.org/10.3390/genes15080996
Bortoletto E, Rosani U. Bioinformatics for Inosine: Tools and Approaches to Trace This Elusive RNA Modification. Genes. 2024; 15(8):996. https://doi.org/10.3390/genes15080996
Chicago/Turabian StyleBortoletto, Enrico, and Umberto Rosani. 2024. "Bioinformatics for Inosine: Tools and Approaches to Trace This Elusive RNA Modification" Genes 15, no. 8: 996. https://doi.org/10.3390/genes15080996
APA StyleBortoletto, E., & Rosani, U. (2024). Bioinformatics for Inosine: Tools and Approaches to Trace This Elusive RNA Modification. Genes, 15(8), 996. https://doi.org/10.3390/genes15080996