
Genomic Regions Associated with Growth and Reproduction Traits in Pink-Eyed White Mink
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Concerns
2.2. Animals and Phenotypes
2.3. Library Construction and Variant Calling
2.4. Population Genome Analysis and Relationship Analysis
2.5. GWAS
2.6. LD and Haplotype Block Analysis
3. Results
3.1. Statistical Description
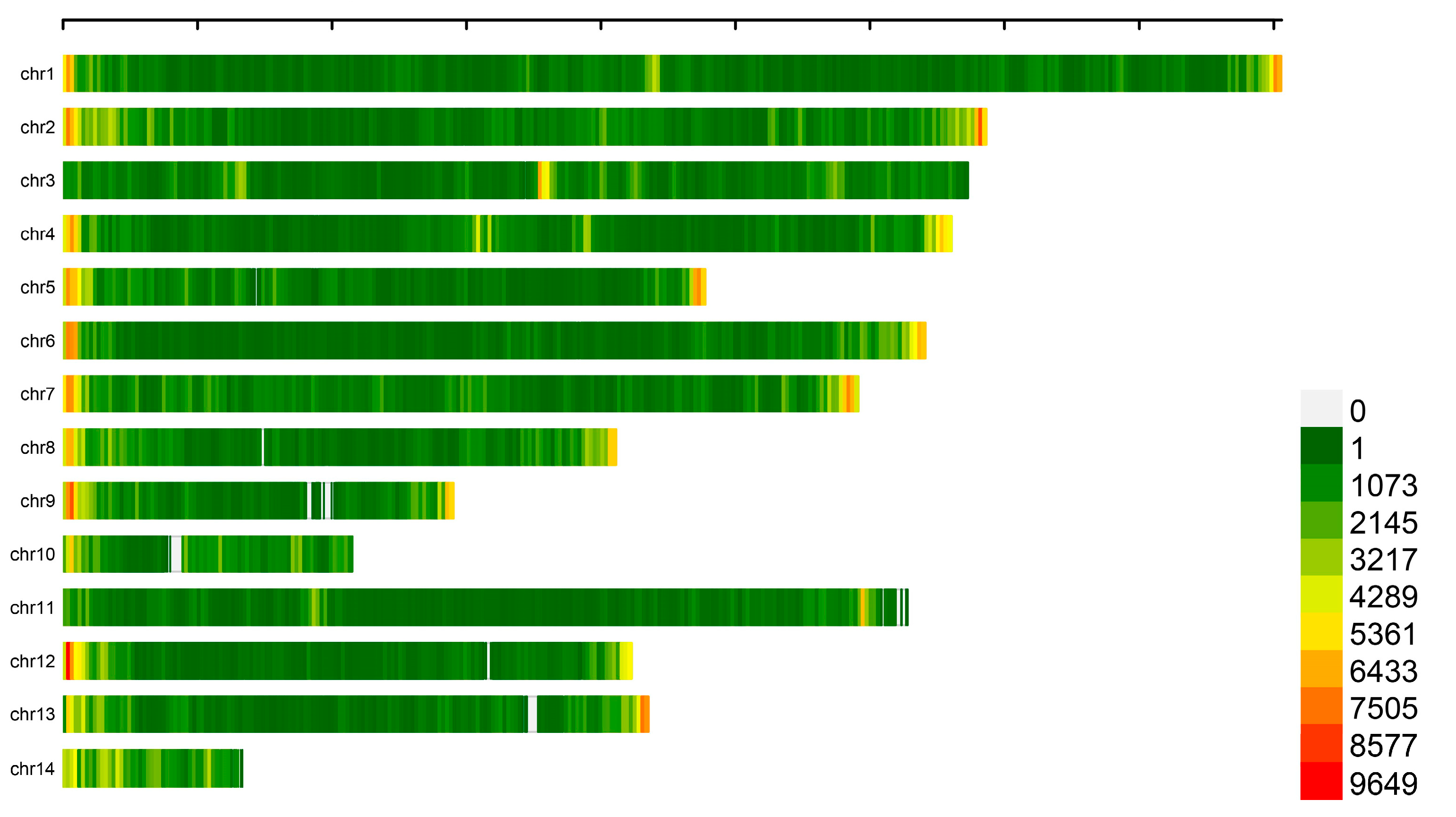
3.2. SNP and Indel Calling
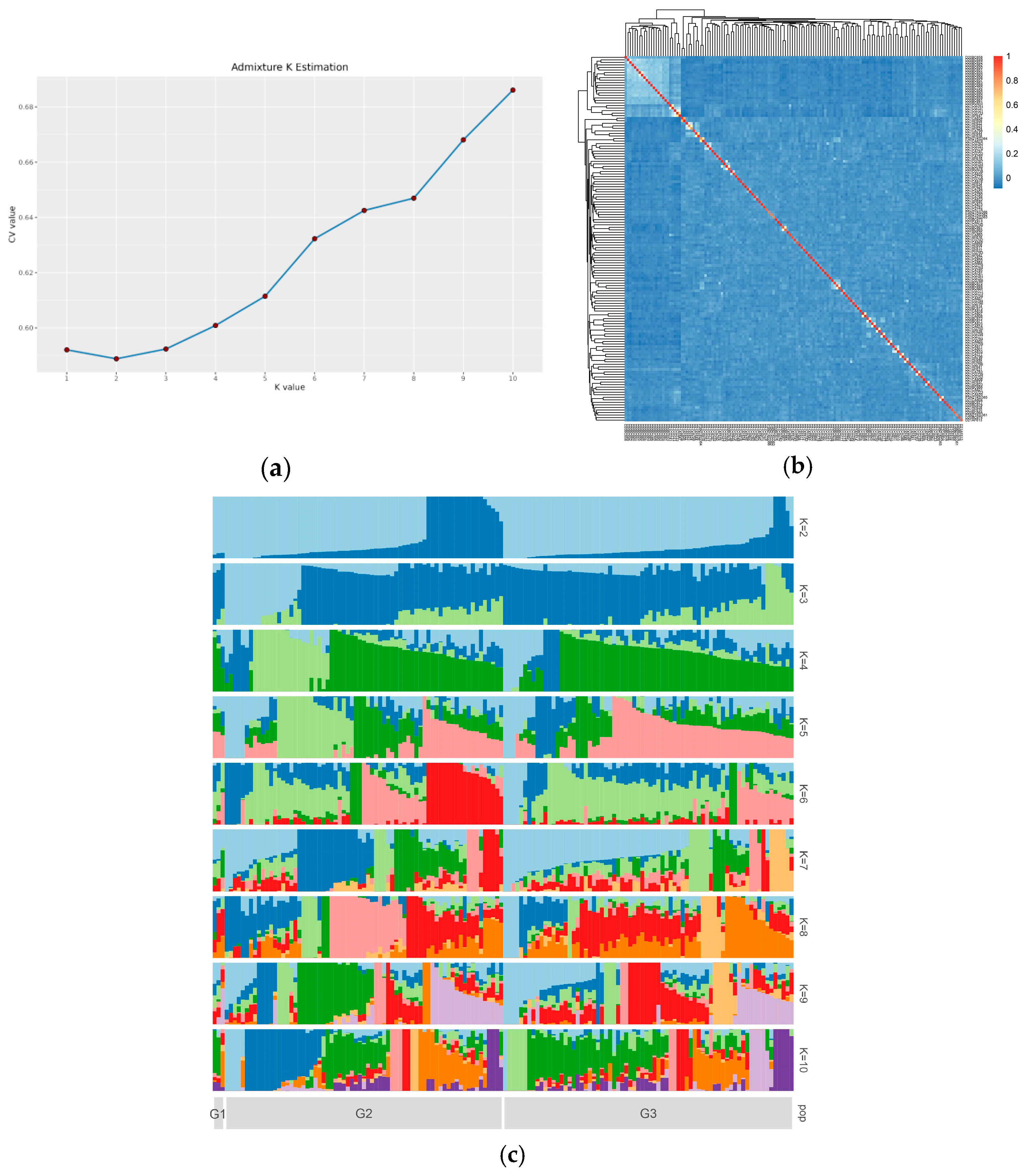
3.3. Population Structures
3.4. Significant SNPs and Genes from the GWAS
3.5. LD and Haplotype Block Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, L.; Zhou, S.; Lyu, T.; Shi, L.; Dong, Y.; He, S.; Zhang, H. Comparative Genome Analysis Reveals the Genomic Basis of Semi-Aquatic Adaptation in American Mink (Neovison Vison). Animals 2022, 12, 2385. [Google Scholar] [CrossRef] [PubMed]
- Manakhov, A.D.; Andreeva, T.V.; Trapezov, O.V.; Kolchanov, N.A.; Rogaev, E.I. Genome Analysis Identifies the Mutant Genes for Common Industrial Silverblue and Hedlund White Coat Colours in American Mink. Sci. Rep. 2019, 9, 4581. [Google Scholar] [CrossRef]
- Markakis, M.N.; Soedring, V.E.; Dantzer, V.; Christensen, K.; Anistoroaei, R. Association of MITF Gene with Hearing and Pigmentation Phenotype in Hedlund White American Mink (Neovison Vison). J. Genet. 2014, 93, 477–481. [Google Scholar] [CrossRef]
- Nes, N.; Einarsson, E.J.; Lohi, O.; Jorgensen, G. Beautiful Fur Animals and Their Colour Genetics; Scientific: Copenhagen, Denmark, 1988. [Google Scholar]
- Davoudi, P.; Do, D.N.; Rathgeber, B.; Colombo, S.M.; Sargolzaei, M.; Plastow, G.; Wang, Z.; Karimi, K.; Hu, G.; Valipour, S.; et al. Genome-Wide Detection of Copy Number Variation in American Mink Using Whole-Genome Sequencing. BMC Genom. 2022, 23, 649. [Google Scholar] [CrossRef]
- Hu, G.; Do, D.N.; Manafiazar, G.; Kelvin, A.A.; Sargolzaei, M.; Plastow, G.; Wang, Z.; Miar, Y. Population Genomics of American Mink Using Genotype Data. Front. Genet. 2023, 14, 1175408. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Villumsen, T.M.; Asp, T.; Guldbrandtsen, B.; Sahana, G.; Lund, M.S. SNP Markers Associated with Body Size and Pelt Length in American Mink (Neovison Vison). BMC Genet. 2018, 19, 103. [Google Scholar] [CrossRef]
- Valipour, S.; Karimi, K.; Do, D.N.; Barrett, D.; Sargolzaei, M.; Plastow, G.; Wang, Z.; Miar, Y. Genome-Wide Detection of Selection Signatures for Pelt Quality Traits and Coat Color Using Whole-Genome Sequencing Data in American Mink. Genes 2022, 13, 1939. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Kai, W.; Li, M.; Hakon, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance Component Model to Account for Sample Structure in Genome-Wide Association Studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef]
- China Commission on Livestock and Poultry Genetic Resources. Animal Genetic Resources in China. Other Animals; China Agriculture Press: Beijing, China, 2012. [Google Scholar]
- Zongyue, L.; Hu, L.; Xiqun, S.; Xiang, L.; Haihua, Z.; Erjun, R.; Baodong, W.; Tietao, Z. Research Progress on Healthy Feeding and Animal Welfare of Mink. J. Econ. Anim. 2023, 1–7. [Google Scholar]
- Karimi, K.; Farid, A.H.; Sargolzaei, M.; Myles, S.; Miar, Y. Linkage Disequilibrium, Effective Population Size and Genomic Inbreeding Rates in American Mink Using Genotyping-by-Sequencing Data. Front. Genet. 2020, 11, 223. [Google Scholar] [CrossRef]
- Zhang, T.; Li, H.; Larsen, P.F.; Ba, H.; Shi, H.; Zhang, H.; Liu, Z. The Genetic Diversity of Mink (Neovison Vison) Populations in China. Animals 2023, 13, 1497. [Google Scholar] [CrossRef] [PubMed]
- Bahl, M.I.; Honoré, A.L.; Skønager, S.T.; Honoré, O.L.; Clausen, T.; Andresen, L.; Hammer, A.S. The Microbiota of Farmed Mink (Neovison Vison) Follows a Successional Development and Is Affected by Early Life Antibiotic Exposure. Sci. Rep. 2020, 10, 20434. [Google Scholar] [CrossRef]
- Karimi, K.; Ngoc Do, D.; Sargolzaei, M.; Miar, Y. Population Genomics of American Mink Using Whole Genome Sequencing Data. Genes 2021, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Lagerkvist, G.; Johansson, K.; Lundeheim, N. Selection for Litter Size, Body Weight, and Pelt Quality in Mink (Mustela Vison): Experimental Design and Direct Response of Each Trait. J. Anim. Sci. 1993, 71, 3261–3272. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.N.; Olesen, C.R.; Hansen, O.; Wamberg, S. Nursing Sickness in Lactating Mink (Mustela Vison). I. Epidemiological and Pathological Observations. Can. J. Vet. Res. 1992, 56, 89–94. [Google Scholar]
- Liu, Z.; Ning, F.; Du, Z.; Yang, C.; Fu, J.; Wang, X.; Bai, X. Modelling Growth of Five Different Colour Types of Mink. SA J. An. Sci. 2011, 41, 116–125. [Google Scholar] [CrossRef]
- Sørensen, K.; Grossman, M.; Koops, W.J. Multiphasic Growth Curves in Mink (Mustela Vison) Selected for Feed Efficiency. Acta Agric. Scand. Sect. A Anim. Sci. 2003, 53, 41–50. [Google Scholar] [CrossRef]
- Mudassir, B.U.; Agha, Z. Microcephaly, Short Stature, Intellectual Disability, Speech Absence and Cataract Are Associated with Novel Bi-Allelic Missense Variant in RTTN Gene: A Seckel Syndrome Case Report. Children 2023, 10, 1027. [Google Scholar] [CrossRef]
- Agnew, T.; Munnur, D.; Crawford, K.; Palazzo, L.; Mikoč, A.; Ahel, I. MacroD1 Is a Promiscuous ADP-Ribosyl Hydrolase Localized to Mitochondria. Front. Microbiol. 2018, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Feijs, K.L.H.; Cooper, C.D.O.; Žaja, R. The Controversial Roles of ADP-Ribosyl Hydrolases MACROD1, MACROD2 and TARG1 in Carcinogenesis. Cancers 2020, 12, 604. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Tauber, M.; Hoybye, C. Endocrine disorders in Prader-Willi syndrome: A model to understand and treat hypothalamic dysfunction. Lancet Diabetes Endocrinol. 2021, 9, 235–246. [Google Scholar] [CrossRef]
- Pacoricona Alfaro, D.L.; Lemoine, P.; Ehlinger, V.; Molinas, C.; Diene, G.; Valette, M.; Pinto, G.; Coupaye, M.; Poitou-Bernert, C.; Thuilleaux, D.; et al. Causes of Death in Prader-Willi Syndrome: Lessons from 11 Years’ Experience of a National Reference Center. Orphanet. J. Rare Dis. 2019, 14, 238. [Google Scholar] [CrossRef]
- Clerc, A.; Coupaye, M.; Mosbah, H.; Pinto, G.; Laurier, V.; Mourre, F.; Merrien, C.; Diene, G.; Poitou, C.; Tauber, M. Diabetes Mellitus in Prader-Willi Syndrome: Natural History during the Transition from Childhood to Adulthood in a Cohort of 39 Patients. J. Clin. Med. 2021, 10, 5310. [Google Scholar] [CrossRef]
- Pascut, D.; Giraudi, P.J.; Banfi, C.; Ghilardi, S.; Tiribelli, C.; Bondesan, A.; Caroli, D.; Minocci, A.; Grugni, G.; Sartorio, A. Proteome Profiling Identifies Circulating Biomarkers Associated with Hepatic Steatosis in Subjects with Prader-Willi Syndrome. Front. Endocrinol. 2023, 14, 1254778. [Google Scholar] [CrossRef]
- Gautason, K. Genomic Selection in Mink Derivation of Economic Values and a Cost-Benefit Analysis of Genomic Selection in Mink Production; Aarhus University: Aarhus, Denmark, 2017. [Google Scholar]
- Clausen, T.N.; Wamberg, S.; Hansen, O. Incidence of Nursing Sickness and Biochemical Observations in Lactating Mink with and without Dietary Salt Supplementation. Can. J. Vet. Res. 1996, 60, 271–276. [Google Scholar]
- Felska-Błaszczyk, L.; Seremak, B. Body Condition of Female Mink (Neovison Vison) Affects Reproduction Performance and Behavior in Pregnancy and Nursing. Acta Sci. Pol. Zootech. 2021, 19, 23–30. [Google Scholar] [CrossRef]
- Karimi, K.; Sargolzaei, M.; Plastow, G.S.; Wang, Z.; Miar, Y. Genetic and Phenotypic Parameters for Litter Size, Survival Rate, Gestation Length, and Litter Weight Traits in American Mink. J. Anim. Sci. 2018, 96, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Wang, Y.; Zhong, L.; Shi, B.; Liang, H.; Han, J. Slc25a36 Modulates Pluripotency of Mouse Embryonic Stem Cells by Regulating Mitochondrial Function and Glutathione Level. Biochem. J. 2019, 476, 1585–1604. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Deng, Q.; Qi, S.; Zhang, Y.; Li, Z.; Yin, S.; Li, Y.; Tan, H.; Wu, M.; Yin, Y.; et al. Genome-Wide Association Analysis Identified Variants Associated with Body Measurement and Reproduction Traits in Shaziling Pigs. Genes 2023, 14, 522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, M.; Yu, R.; Liu, B.; Tian, Z.; Liu, S. FSCB Phosphorylation Regulates Mouse Spermatozoa Capacitation through Suppressing SUMOylation of ROPN1/ROPN1L. Am. J. Transl. Res. 2016, 8, 2776–2782. [Google Scholar]
- Shorten, P.R.; Ledgard, A.M.; Donnison, M.; Pfeffer, P.L.; McDonald, R.M.; Berg, D.K. A Mathematical Model of the Interaction between Bovine Blastocyst Developmental Stage and Progesterone-Stimulated Uterine Factors on Differential Embryonic Development Observed on Day 15 of Gestation. J. Dairy Sci. 2018, 101, 736–751. [Google Scholar] [CrossRef]
- Nayeri, S.; Sargolzaei, M.; Abo-Ismail, M.K.; May, N.; Miller, S.P.; Schenkel, F.; Moore, S.S.; Stothard, P. Genome-Wide Association for Milk Production and Female Fertility Traits in Canadian Dairy Holstein Cattle. BMC Genet. 2016, 17, 75. [Google Scholar] [CrossRef]
- Illa, S.K.; Mukherjee, S.; Nath, S.; Mukherjee, A. Genome-Wide Scanning for Signatures of Selection Revealed the Putative Genomic Regions and Candidate Genes Controlling Milk Composition and Coat Color Traits in Sahiwal Cattle. Front. Genet. 2021, 12, 699422. [Google Scholar] [CrossRef]
- Tek, B.; Elçin, P.; Tunçdemir, M.; Onaran, İ.; Özkara, H.; Kanıgür Sultuybek, G. A Role for Heterozygosity of NF-κB1 Rs28362491 Polymorphism in Patients with Idiopathic Oligospermia. Arch. Iran Med. 2016, 19, 275–281. [Google Scholar]
- Wang, T.; Hu, T.; Zhen, J.; Zhang, L.; Zhang, Z. Association of MTHFR, NFKB1, NFKBIA, DAZL and CYP1A1 Gene Polymorphisms with Risk of Idiopathic Male Infertility in a Han Chinese Population. Int. J. Clin. Exp. Pathol. 2017, 10, 7640–7649. [Google Scholar]
- Vangeel, E.B.; Pishva, E.; Hompes, T.; van den Hove, D.; Lambrechts, D.; Allegaert, K.; Freson, K.; Izzi, B.; Claes, S. Newborn Genome-Wide DNA Methylation in Association with Pregnancy Anxiety Reveals a Potential Role for GABBR1. Clin. Epigenetics 2017, 9, 107. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Fixed Effects | λ | |||||
|---|---|---|---|---|---|---|---|
| Sex | Age | Length of Gestation | PCA1 | PCA2 | PCA3 | ||
| TNB | √ | √ | √ | √ | √ | √ | 0.940 |
| NBA | √ | √ | √ | √ | √ | √ | 0.961 |
| BW | √ | √ | √ | √ | √ | 0.956 | |
| Traits | Female | Male | ||
|---|---|---|---|---|
| TNB | NBA | BW | BW | |
| Mean ± SD | 6.45 ± 2.08 | 5.17 ± 1.71 | 1.65 ± 0.28 | 3.14 ± 0.31 |
| Max | 11.00 | 9.00 | 2.2 | 3.70 |
| Min | 2.00 | 0.00 | 1.04 | 2.42 |
| Types | Total | |
|---|---|---|
| Number | % | |
| Exonic total | 67,192 | 2.78 |
| Synonymous | 34,431 | 1.43 |
| Non-synonymous | 30,498 | 1.26 |
| Stop-gain | 777 | 0.03 |
| Stop-loss | 35 | 0 |
| Unknown | 1451 | 0.06 |
| Splicing | 440 | 0.02 |
| ncRNA total | 44 | 0 |
| ncRNA_exonic | 44 | 0 |
| ncRNA_splicing | 0 | 0 |
| ncRNA_exonic; splicing | 0 | 0 |
| ncRNA_intronic | 0 | 0 |
| Intronic | 1,019,723 | 42.22 |
| Intergenic | 1,238,143 | 51.27 |
| UTR5 | 7589 | 0.31 |
| UTR3 | 27,628 | 1.14 |
| UTR5;UTR3 | 2 | 0 |
| Upstream | 26,832 | 1.11 |
| Downstream | 26,171 | 1.08 |
| Upstream; downstream | 1357 | 0.06 |
| Total | 2,415,121 | 100 |
| Traits | Chr | Position | MAF | log10(P) | Genes |
|---|---|---|---|---|---|
| NBA | 6 | 67,634,385 | 0.142 | 6.0725631 | STXBP5L, SLC25A36 |
| 13 | 49,926,996 | 0.1562 | 7.1691173 | RPS29, LOC122893428 | |
| TNB | 1 | 13,908,286 | 0.1114 | 5.0168059 | ATXN1 (intronic) |
| 1 | 15,071,975 | 0.0369 | 5.2790484 | GABBR1 (521753) | |
| 2 | 9,564,047 | 0.3286 | 5.0929676 | PDPN (intronic) | |
| 2 | 12,429,330 | 0.1337 | 5.0591373 | ARHGEF10L (intronic) | |
| 4 | 21,260,036 | 0.1108 | 5.1866097 | SAMD12 (intronic) | |
| 13 | 49,926,996 | 0.1562 | 5.6864521 | RPS29 (629,685), LOC122893428 (1,047,595) | |
| 13 | 54,658,193 | 0.1628 | 5.1395122 | FSCB (771,634), LRFN5 (892,149) | |
| 13 | 54,658,194 | 0.1628 | 5.1395122 | FSCB (771,635), LRFN5 (1,892,148) | |
| 13 | 54,658,196 | 0.1628 | 5.1395122 | FSCB (771,637), LRFN5 (1,892,146) | |
| 13 | 54,658,197 | 0.1628 | 5.1395122 | FSCB (771,638), LRFN5 (1,892,145) | |
| 13 | 61,338,532 | 0.0909 | 5.4076409 | NKX2-1 (61,961), LOC122894326 (104,961) | |
| 13 | 62,308,394 | 0.0966 | 5.1340934 | NFKBIA (28,135), PSMA6 (32,817) | |
| 13 | 65,124,124 | 0.1733 | 5.0066897 | AKAP6 (intronic) | |
| BW | 3 | 13,116,054 | 0.3636 | 5.2561596 | RTTN (intronic) |
| 7 | 45,758,732 | 0.05398 | 5.0827756 | PRPF31 (exonic) | |
| 7 | 19,922,808 | 0.2861 | 5.0507150 | MACROD1 (intronic) | |
| 9 | 6,802,276 | 0.05398 | 5.3573332 | KYAT1 (UTR3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, H.; Liu, L.; Larsen, P.F.; Ding, Y.; Zhang, T.; Zhang, H.; Liu, Z. Genomic Regions Associated with Growth and Reproduction Traits in Pink-Eyed White Mink. Genes 2024, 15, 1142. https://doi.org/10.3390/genes15091142
Shi H, Liu L, Larsen PF, Ding Y, Zhang T, Zhang H, Liu Z. Genomic Regions Associated with Growth and Reproduction Traits in Pink-Eyed White Mink. Genes. 2024; 15(9):1142. https://doi.org/10.3390/genes15091142
Chicago/Turabian StyleShi, Hongyu, Linling Liu, Peter Foged Larsen, Yu Ding, Tietao Zhang, Haihua Zhang, and Zongyue Liu. 2024. "Genomic Regions Associated with Growth and Reproduction Traits in Pink-Eyed White Mink" Genes 15, no. 9: 1142. https://doi.org/10.3390/genes15091142