
RHO Variants and Autosomal Dominant Retinitis Pigmentosa: Insights from the Italian Genetic Landscape
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cohort Description
2.2. DNA Purification and Quantification
2.3. Whole Exome Sequencing (WES) and Variant Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strafella, C.; Caputo, V.; Pagliaroli, G.; Iozzo, N.; Campoli, G.; Carboni, S.; Peconi, C.; Galota, R.M.; Zampatti, S.; Minozzi, G.; et al. NGS Analysis for Molecular Diagnosis of Retinitis Pigmentosa (RP): Detection of a Novel Variant in PRPH2 Gene. Genes 2019, 10, 792. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013, 84, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Retinitis Pigmentosa (Non-syndromic). In Atlas of Inherited Retinal Diseases; Tsang, S.H., Sharma, T., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 125–130. ISBN 978-3-319-95046-4. [Google Scholar]
- Bhardwaj, A.; Yadav, A.; Yadav, M.; Tanwar, M. Genetic dissection of non-syndromic retinitis pigmentosa. Indian J. Ophthalmol. 2022, 70, 2355. [Google Scholar] [CrossRef]
- Chang, S.; Vaccarella, L.; Olatunji, S.; Cebulla, C.; Christoforidis, J. Diagnostic Challenges in Retinitis Pigmentosa: Genotypic Multiplicity and Phenotypic Variability. Curr. Genom. 2011, 12, 267–275. [Google Scholar] [CrossRef]
- Kim, C.; Kim, K.J.; Bok, J.; Lee, E.-J.; Kim, D.-J.; Oh, J.H.; Park, S.P.; Shin, J.Y.; Lee, J.-Y.; Yu, H.G. Microarray-based mutation detection and phenotypic characterization in Korean patients with retinitis pigmentosa. Mol. Vis. 2012, 18, 2398–2410. [Google Scholar]
- Wu, J.; Chen, L.; Tam, O.S.; Huang, X.-F.; Pang, C.-P.; Jin, Z.-B. Whole Exome Sequencing Reveals Genetic Predisposition in a Large Family with Retinitis Pigmentosa. BioMed Res. Int. 2014, 2014, 302487. [Google Scholar] [CrossRef]
- Wu, Y.; Guo, Y.; Yi, J.; Xu, H.; Yuan, L.; Yang, Z.; Deng, H. Heterozygous RHO p.R135W missense mutation in a large Han-Chinese family with retinitis pigmentosa and different refractive errors. Biosci. Rep. 2019, 39, BSR20182198. [Google Scholar] [CrossRef]
- Wang, D.Y.; Chan, W.M.; Tam, P.O.S.; Baum, L.; Lam, D.S.C.; Chong, K.K.L.; Fan, B.J.; Pang, C.P. Gene mutations in retinitis pigmentosa and their clinical implications. Clin. Chim. Acta 2005, 351, 5–16. [Google Scholar] [CrossRef]
- Colombo, L.; Maltese, P.E.; Castori, M.; El Shamieh, S.; Zeitz, C.; Audo, I.; Zulian, A.; Marinelli, C.; Benedetti, S.; Costantini, A.; et al. Molecular Epidemiology in 591 Italian Probands with Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Investig. Ophthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.H.; Wensel, T.G. The nature of dominant mutations of rhodopsin and implications for gene therapy. Mol. Neurobiol. 2003, 28, 149–158. [Google Scholar] [CrossRef]
- Malanson, K.M.; Lem, J. Chapter 1 Rhodopsin-Mediated Retinitis Pigmentosa. In Progress in Molecular Biology and Translational Science; G Protein-Coupled Receptors in Health and Disease, Part A; Academic Press: Cambridge, MA, USA, 2009; Volume 88, pp. 1–31. [Google Scholar]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis Pigmentosa: Genes and Disease Mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Opefi, C.A.; South, K.; Bellingham, J.; Bevilacqua, D.; Munro, P.M.; Kanuga, N.; Mackenzie, F.E.; Dubis, A.M.; et al. Rescue of mutant rhodopsin traffic by metformin-induced AMPK activation accelerates photoreceptor degeneration. Hum. Mol. Genet. 2017, 26, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liu, Z.-L.; Zhang, X.; Wang, W.; Huang, Z.-Q.; Sun, S.-N.; Jin, Z.-B. Modeling autosomal dominant retinitis pigmentosa by using patient-specific retinal organoids with a class-3 RHO mutation. Exp. Eye Res. 2024, 241, 109856. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Zhen, F.; Zou, T.; Wang, T.; Zhou, Y.; Dong, S.; Zhang, H. Rhodopsin-associated retinal dystrophy: Disease mechanisms and therapeutic strategies. Front. Neurosci. 2023, 17, 1132179. [Google Scholar] [CrossRef]
- Singhal, A.; Ostermaier, M.K.; Vishnivetskiy, S.A.; Panneels, V.; Homan, K.T.; Tesmer, J.J.G.; Veprintsev, D.; Deupi, X.; Gurevich, V.V.; Schertler, G.F.X.; et al. Insights into congenital stationary night blindness based on the structure of G90D rhodopsin. EMBO Rep. 2013, 14, 520–526. [Google Scholar] [CrossRef]
- Kumaramanickavel, G.; Maw, M.; Denton, M.J.; John, S.; Srikumari, C.R.S.; Orth, U.; Oehlmann, R.; Gal, A. Missense rhodopsin mutation in a family with recessive RP. Nat. Genet. 1994, 8, 10–11. [Google Scholar] [CrossRef]
- Kartasasmita, A.; Fujiki, K.; Iskandar, E.; Sovani, I.; Fujimaki, T.; Murakami, A. A novel nonsense mutation in Rhodopsin gene in two Indonesian Families with Autosomal Recessive Retinitis Pigmentosa. Ophthalmic Genet. 2011, 32, 57–63. [Google Scholar] [CrossRef]
- Azam, M.; Khan, M.I.; Gal, A.; Hussain, A.; Shah, S.T.A.; Khan, M.S.; Sadeque, A.; Bokhari, H.; Collin, R.W.J.; Orth, U.; et al. A homozygous p.Glu150Lys mutation in the opsin gene of two Pakistani families with autosomal recessive retinitis pigmentosa. Mol. Vis. 2009, 15, 2526–2534. [Google Scholar] [PubMed]
- Luo, H.; Xiao, X.; Li, S.; Sun, W.; Yi, Z.; Wang, P.; Zhang, Q. Spectrum-frequency and genotype–phenotype analysis of rhodopsin variants. Exp. Eye Res. 2021, 203, 108405. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, P.J.; Cowley, G.S.; McGee, T.L.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. A Null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat. Genet. 1992, 1, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Lu, T.; Zhang, X.; Dai, H.; Yan, W.; Bai, F.; Li, Y. Identification of two mutations of the RHO gene in two Chinese families with retinitis pigmentosa: Correlation between genotype and phenotype. Mol. Vis. 2012, 18, 3013–3020. [Google Scholar] [PubMed]
- Zhang, X.L.; Liu, M.; Meng, X.H.; Fu, W.L.; Yin, Z.Q.; Huang, J.F.; Zhang, X. Mutational analysis of the rhodopsin gene in Chinese ADRP families by conformation sensitive gel electrophoresis. Life Sci. 2006, 78, 1494–1498. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Ellard, S.; Baple, E.L.; Callaway, A.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020. Available online: https://www.semanticscholar.org/paper/ACGS-Best-Practice-Guidelines-for-Variant-in-Rare-Ellard-Baple/7923ccd6fdd28c82e48f05f5117d8bd261d180d9 (accessed on 21 July 2024).
- Lim, K.P.; Yip, S.P.; Cheung, S.C.; Leung, K.W.; Lam, S.T.S.; To, C.H. Novel PRPF31 and PRPH2 Mutations and Co-occurrence of PRPF31 and RHO Mutations in Chinese Patients with Retinitis Pigmentosa. Arch. Ophthalmol. 2009, 127, 784–790. [Google Scholar] [CrossRef]
- Tsutsui, S.; Murakami, Y.; Fujiwara, K.; Koyanagi, Y.; Akiyama, M.; Takeda, A.; Ikeda, Y.; Sonoda, K.-H. Genotypes and clinical features of RHO-associated retinitis pigmentosa in a Japanese population. Jpn. J. Ophthalmol. 2024, 68, 1–11. [Google Scholar] [CrossRef]
- Kučinskas, V.; Payne, A.M.; Ambrasienė, D.; Jurgelevičius, V.; Steponavičiūtė, D.; Arčiulienė, J.; Daktaravičienė, E.; Bhattacharya, S. Molecular Genetic Study of Autosomal Dominant Retinitis pigmentosa in Lithuanian Patients. Hum. Hered. 1999, 49, 71–74. [Google Scholar] [CrossRef]
- Li, S.; Xiao, X.; Wang, P.; Guo, X.; Zhang, Q. Mutation spectrum and frequency of the RHO gene in 248 Chinese families with retinitis pigmentosa. Biochem. Biophys. Res. Commun. 2010, 401, 42–47. [Google Scholar] [CrossRef]
- Fernandez-San Jose, P.; Blanco-Kelly, F.; Corton, M.; Trujillo-Tiebas, M.-J.; Gimenez, A.; Avila-Fernandez, A.; Garcia-Sandoval, B.; Lopez-Molina, M.-I.; Hernan, I.; Carballo, M.; et al. Prevalence of Rhodopsin mutations in autosomal dominant Retinitis Pigmentosa in Spain: Clinical and analytical review in 200 families. Acta Ophthalmol. 2015, 93, e38–e44. [Google Scholar] [CrossRef]
- Massof, R.W.; Finkelstein, D. Vision Threshold Profiles in Sector Retinitis Pigmentosa. Arch. Ophthalmol. 1979, 97, 1899–1904. [Google Scholar] [CrossRef]
- Zhuang, J.; Zhang, R.; Zhou, B.; Cao, Z.; Zhou, J.; Chen, X.; Zhang, N.; Zhu, Y.; Yang, J. Mutation analysis of RHO in patients with non-syndromic retinitis pigmentosa. Ophthalmic Genet. 2024, 45, 147–152. [Google Scholar] [CrossRef]
- Concepcion, F.; Chen, J. Q344ter Mutation Causes Mislocalization of Rhodopsin Molecules That Are Catalytically Active: A Mouse Model of Q344ter-Induced Retinal Degeneration. PLoS ONE 2010, 5, e10904. [Google Scholar] [CrossRef]
- Yong, R.; Chee, C.; Yap, E. A Two-stage Approach Identifies a Q344X Mutation in the Rhodopsin Gene of a Chinese Singaporean Family with Autosomal Dominant Retinitis Pigmentosa. Ann. Acad. Med. Singap. 2005, 34, 94–99. [Google Scholar] [CrossRef]
- Schorderet, D.F.; Iouranova, A.; Favez, T.; Tiab, L.; Escher, P. IROme, a New High-Throughput Molecular Tool for the Diagnosis of Inherited Retinal Dystrophies. BioMed Res. Int. 2013, 2013, 198089. [Google Scholar] [CrossRef]
- Ma, D.J.; Lee, H.-S.; Kim, K.; Choi, S.; Jang, I.; Cho, S.-H.; Yoon, C.K.; Lee, E.K.; Yu, H.G. Whole-exome sequencing in 168 Korean patients with inherited retinal degeneration. BMC Med. Genom. 2021, 14, 74. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.P.; Holden, D.C.; Joshi, P.; Clark, C.L.; Lee, A.H.; Kaushal, S. Molecular Mechanisms of Rhodopsin Retinitis Pigmentosa and the Efficacy of Pharmacological Rescue. J. Mol. Biol. 2010, 395, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Carrell, R.W. Cell toxicity and conformational disease. Trends Cell Biol. 2005, 15, 574–580. [Google Scholar] [CrossRef]
- Gregersen, N.; Bross, P.; Vang, S.; Christensen, J.H. Protein Misfolding and Human Disease. Annu. Rev. Genom. Hum. Genet. 2006, 7, 103–124. [Google Scholar] [CrossRef]
- Millá, E.; Maseras, M.; Martínez-Gimeno, M.; Gamundi, M.J.; Assaf, H.; Esmerado, C.; Carballo, M.; Grupo Multicéntrico Español de Retinosis. Pigmentaria Genetic and molecular characterization of 148 patients with autosomal dominant retinitis pigmentosa (ADRP). Arch. Soc. Espanola Oftalmol. 2002, 77, 481–484. [Google Scholar]
- Bunge, S.; Wedemann, H.; David, D.; Terwilliger, D.J.; van den Born, L.I.; Aulehla-Scholz, C.; Samanns, C.; Horn, M.; Ott, J.; Schwinger, E.; et al. Molecular Analysis and Genetic Mapping of the Rhodopsin Gene in Families with Autosomal Dominant Retinitis Pigmentosa. Genomics 1993, 17, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Ziviello, C.; Simonelli, F.; Testa, F.; Anastasi, M.; Marzoli, S.B.; Falsini, B.; Ghiglione, D.; Macaluso, C.; Manitto, M.P.; Garrè, C.; et al. Molecular genetics of autosomal dominant retinitis pigmentosa (ADRP): A comprehensive study of 43 Italian families. J. Med. Genet. 2005, 42, e47. [Google Scholar] [CrossRef] [PubMed]
- Bareil, C.; Hamel, C.; Pallarès-Ruiz, N.; Arnaud, B.; Demaille, J.; Claustres, M. Molecular analysis of the rhodopsin gene in southern France: Identification of the first duplication responsible for retinitis pigmentosa, c.998^999ins4. Ophthalmic Genet. 1999, 20, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Sharma, T. Autosomal Dominant Retinitis Pigmentosa. In Atlas of Inherited Retinal Diseases; Tsang, S.H., Sharma, T., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 69–77. ISBN 978-3-319-95046-4. [Google Scholar]
- Pierrottet, C.O.; Zuntini, M.; Digiuni, M.; Bazzanella, I.; Ferri, P.; Paderni, R.; Rossetti, L.M.; Cecchin, S.; Orzalesi, N.; Bertelli, M. Syndromic and non-syndromic forms of retinitis pigmentosa: A comprehensive Italian clinical and molecular study reveals new mutations. Genet. Mol. Res. 2014, 13, 8815–8833. [Google Scholar] [CrossRef]
- Audo, I.; Manes, G.; Mohand-Saïd, S.; Friedrich, A.; Lancelot, M.-E.; Antonio, A.; Moskova-Doumanova, V.; Poch, O.; Zanlonghi, X.; Hamel, C.P.; et al. Spectrum of Rhodopsin Mutations in French Autosomal Dominant Rod–Cone Dystrophy Patients. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3687–3700. [Google Scholar] [CrossRef]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Dryja, T.P. Ocular Findings in Patients with Autosomal Dominant Retinitis Pigmentosa and Rhodopsin, Proline-347-Leucine. Am. J. Ophthalmol. 1991, 111, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Dikshit, M.; Agarwal, R. Mutation analysis of codons 345 and 347 of rhodopsin gene in Indian retinitis pigmentosa patients. J. Genet. 2001, 80, 111–116. [Google Scholar] [CrossRef]
- Yang, G.; Xie, S.; Feng, N.; Yuan, Z.; Zhang, M.; Zhao, J. Spectrum of rhodopsin gene mutations in Chinese patients with retinitis pigmentosa. Mol. Vis. 2014, 20, 1132–1136. [Google Scholar]
- Karali, M.; Testa, F.; Di Iorio, V.; Torella, A.; Zeuli, R.; Scarpato, M.; Romano, F.; Onore, M.E.; Pizzo, M.; Melillo, P.; et al. Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy. Sci. Rep. 2022, 12, 20815. [Google Scholar] [CrossRef]
- Park, S.P.; Lee, W.; Bae, E.J.; Greenstein, V.; Sin, B.H.; Chang, S.; Tsang, S.H. Early Structural Anomalies Observed by High-Resolution Imaging in Two Related Cases of Autosomal-Dominant Retinitis Pigmentosa. Ophthalmic Surg. Lasers Imaging Retin. 2014, 45, 469–473. [Google Scholar] [CrossRef]
- Iannaccone, A.; Man, D.; Waseem, N.; Jennings, B.J.; Ganapathiraju, M.; Gallaher, K.; Reese, E.; Bhattacharya, S.S.; Klein-Seetharaman, J. Retinitis pigmentosa associated with rhodopsin mutations: Correlation between phenotypic variability and molecular effects. Vision Res. 2006, 46, 4556–4567. [Google Scholar] [CrossRef]
- Jung, Y.H.; Kwak, J.J.; Joo, K.; Lee, H.J.; Park, K.H.; Kim, M.S.; Lee, E.K.; Byeon, S.H.; Lee, C.S.; Han, J.; et al. Clinical and genetic features of Koreans with retinitis pigmentosa associated with mutations in rhodopsin. Front. Genet. 2023, 14, 1240067. [Google Scholar] [CrossRef]
- Fernández-Sánchez, L.; Lax, P.; Pinilla, I.; Martín-Nieto, J.; Cuenca, N. Tauroursodeoxycholic Acid Prevents Retinal Degeneration in Transgenic P23H Rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4998–5008. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Jastrzebska, B.; Golczak, M.; Gulati, S.; Tang, H.; Seibel, W.; Li, X.; Jin, H.; Han, Y.; et al. A novel small molecule chaperone of rod opsin and its potential therapy for retinal degeneration. Nat. Commun. 2018, 9, 1976. [Google Scholar] [CrossRef]
- Ahmed, C.M.; Massengill, M.T.; Ildefonso, C.J.; Jalligampala, A.; Zhu, P.; Li, H.; Patel, A.P.; McCall, M.A.; Lewin, A.S. Binocular benefit following monocular subretinal AAV injection in a mouse model of autosomal dominant retinitis pigmentosa (adRP). Vision Res. 2023, 206, 108189. [Google Scholar] [CrossRef]
- Li, P.; Kleinstiver, B.P.; Leon, M.Y.; Prew, M.S.; Navarro-Gomez, D.; Greenwald, S.H.; Pierce, E.A.; Joung, J.K.; Liu, Q. Allele-Specific CRISPR-Cas9 Genome Editing of the Single-Base P23H Mutation for Rhodopsin-Associated Dominant Retinitis Pigmentosa. CRISPR J. 2018, 1, 55–64. [Google Scholar] [CrossRef]
- Wheway, G.; Douglas, A.; Baralle, D.; Guillot, E. Mutation spectrum of PRPF31, genotype-phenotype correlation in retinitis pigmentosa, and opportunities for therapy. Exp. Eye Res. 2020, 192, 107950. [Google Scholar] [CrossRef]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; da Silva Cunha, A.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef]
- Diakatou, M.; Manes, G.; Bocquet, B.; Meunier, I.; Kalatzis, V. Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 20, 2542. [Google Scholar] [CrossRef]
- Tebbe, L.; Kakakhel, M.; Makia, M.S.; Al-Ubaidi, M.R.; Naash, M.I. The Interplay between Peripherin 2 Complex Formation and Degenerative Retinal Diseases. Cells 2020, 9, 784. [Google Scholar] [CrossRef]
- Rutan Woods, C.T.; Makia, M.S.; Lewis, T.R.; Crane, R.; Zeibak, S.; Yu, P.; Kakakhel, M.; Castillo, C.M.; Arshavsky, V.Y.; Naash, M.I.; et al. Downregulation of rhodopsin is an effective therapeutic strategy in ameliorating peripherin-2-associated inherited retinal disorders. Nat. Commun. 2024, 15, 4756. [Google Scholar] [CrossRef]
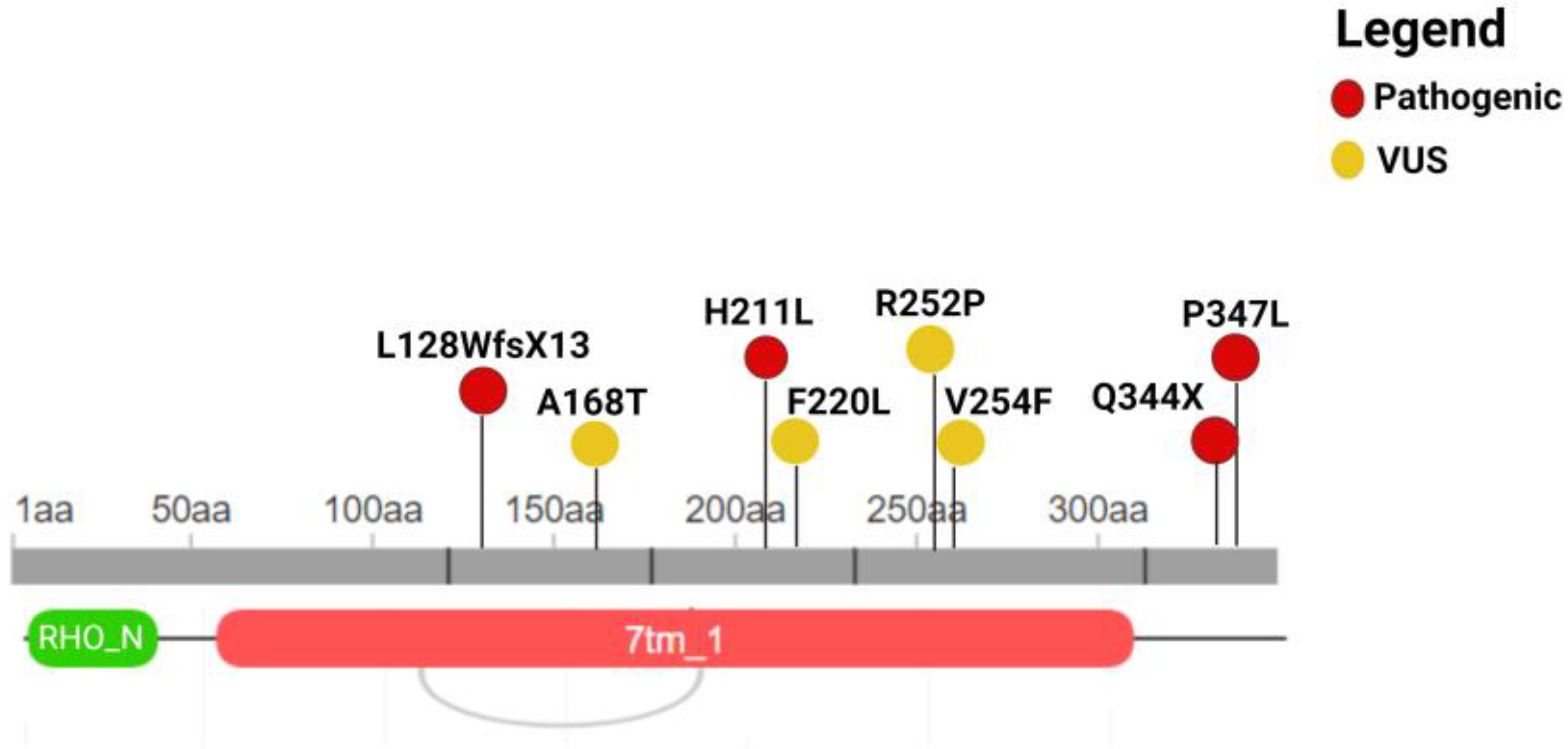

{kind=link}
| Genomic Position | Exon | Nucleotide Coding | Protein Coding | Rs | Effect | ACMG Classification |
|---|---|---|---|---|---|---|
| 3:129251195 | 3 | c.632A>T | p.H211L | Na | missense | Pathogenic |
| 3:129252554 | 5 | c.1040C>T | p.P347L | rs29001566 | missense | Pathogenic |
| 3:129252544 | 5 | c.1030C>T | p.Q344X | rs104893778 | nonsense | Pathogenic |
| 3:129249740_129249749 | 2 | c.383_392del | p.L128WfsX13 | Na | frameshift | Pathogenic |
| 3:129251439 | 4 | c.760G>T | p.V254F | Na | missense | VUS |
| 3:129249859 | 2 | c.502G>A | p.A168T | rs574202023 | missense | VUS |
| 3:129251223 | 3 | c.660T>G | p.F220L | rs141956356 | missense | VUS |
| 3:129251434 | 4 | c.755G>C | p.R252P | rs765438313 | missense | VUS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trastulli, G.; Megalizzi, D.; Calvino, G.; Andreucci, S.; Zampatti, S.; Strafella, C.; Caltagirone, C.; Giardina, E.; Cascella, R. RHO Variants and Autosomal Dominant Retinitis Pigmentosa: Insights from the Italian Genetic Landscape. Genes 2024, 15, 1158. https://doi.org/10.3390/genes15091158
Trastulli G, Megalizzi D, Calvino G, Andreucci S, Zampatti S, Strafella C, Caltagirone C, Giardina E, Cascella R. RHO Variants and Autosomal Dominant Retinitis Pigmentosa: Insights from the Italian Genetic Landscape. Genes. 2024; 15(9):1158. https://doi.org/10.3390/genes15091158
Chicago/Turabian StyleTrastulli, Giulia, Domenica Megalizzi, Giulia Calvino, Sarah Andreucci, Stefania Zampatti, Claudia Strafella, Carlo Caltagirone, Emiliano Giardina, and Raffaella Cascella. 2024. "RHO Variants and Autosomal Dominant Retinitis Pigmentosa: Insights from the Italian Genetic Landscape" Genes 15, no. 9: 1158. https://doi.org/10.3390/genes15091158