
A Variant-Centric Analysis of Allele Sharing in Dogs and Wolves


Abstract
:1. Introduction
2. Materials and Methods
2.1. Canine Variant Data Processing
2.2. SNP Distance Calculation
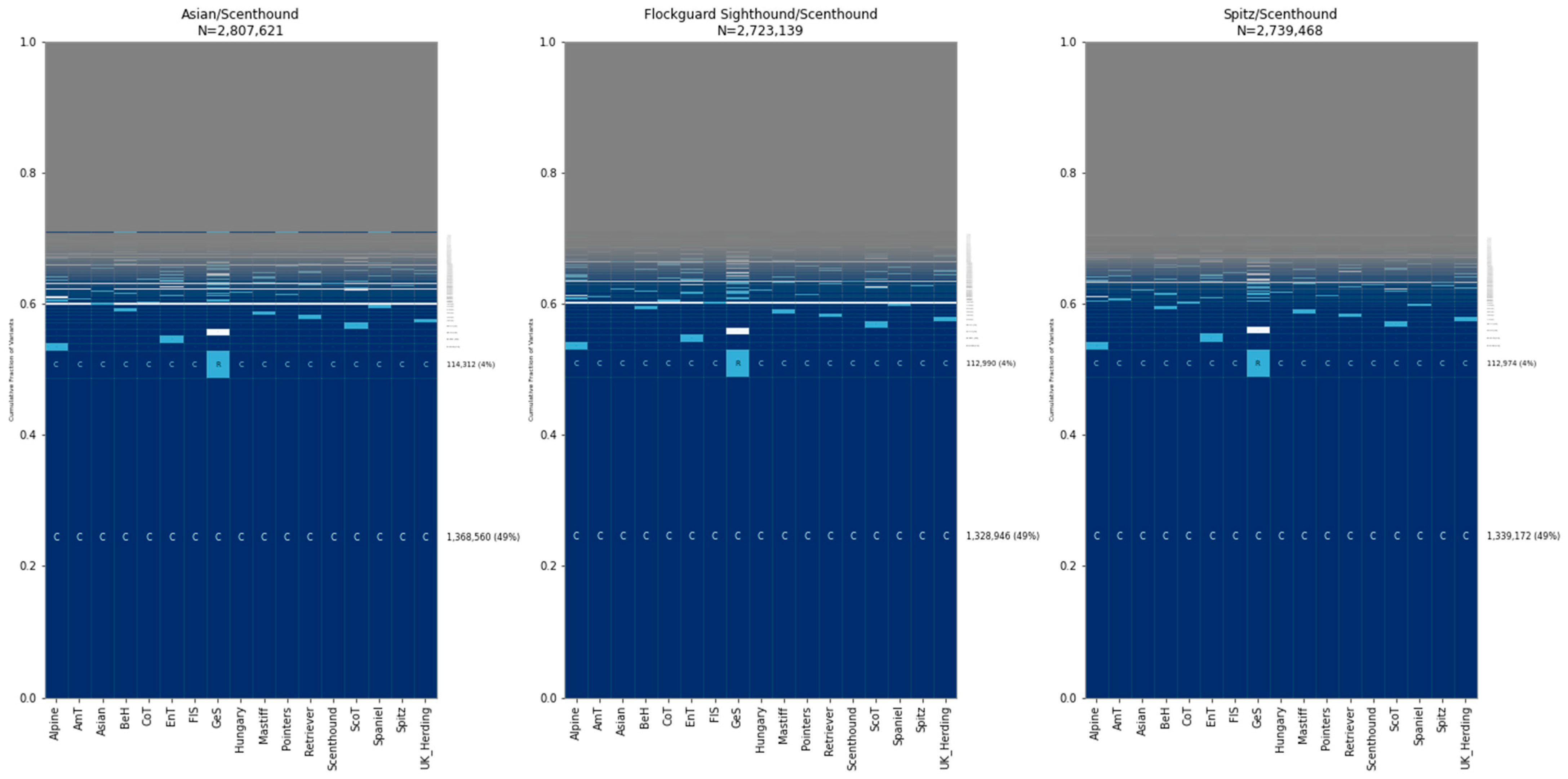
2.3. Canine GeoVar Analysis
2.3.1. Village Dog Classification
2.3.2. Breed Dog Classification
2.3.3. Microarray and Two-Sample Comparisons
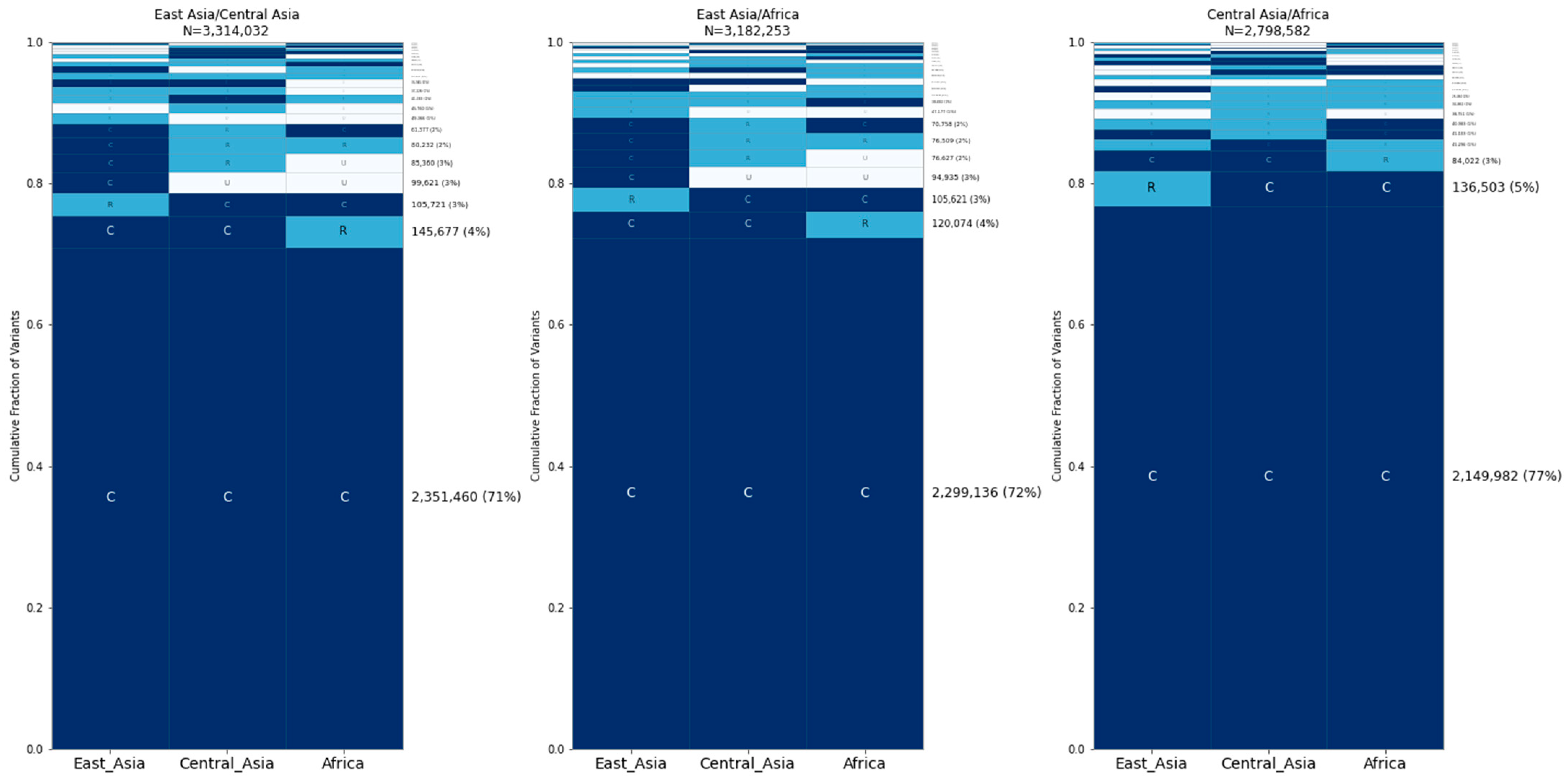
2.4. Human GeoVar Analysis
3. Results
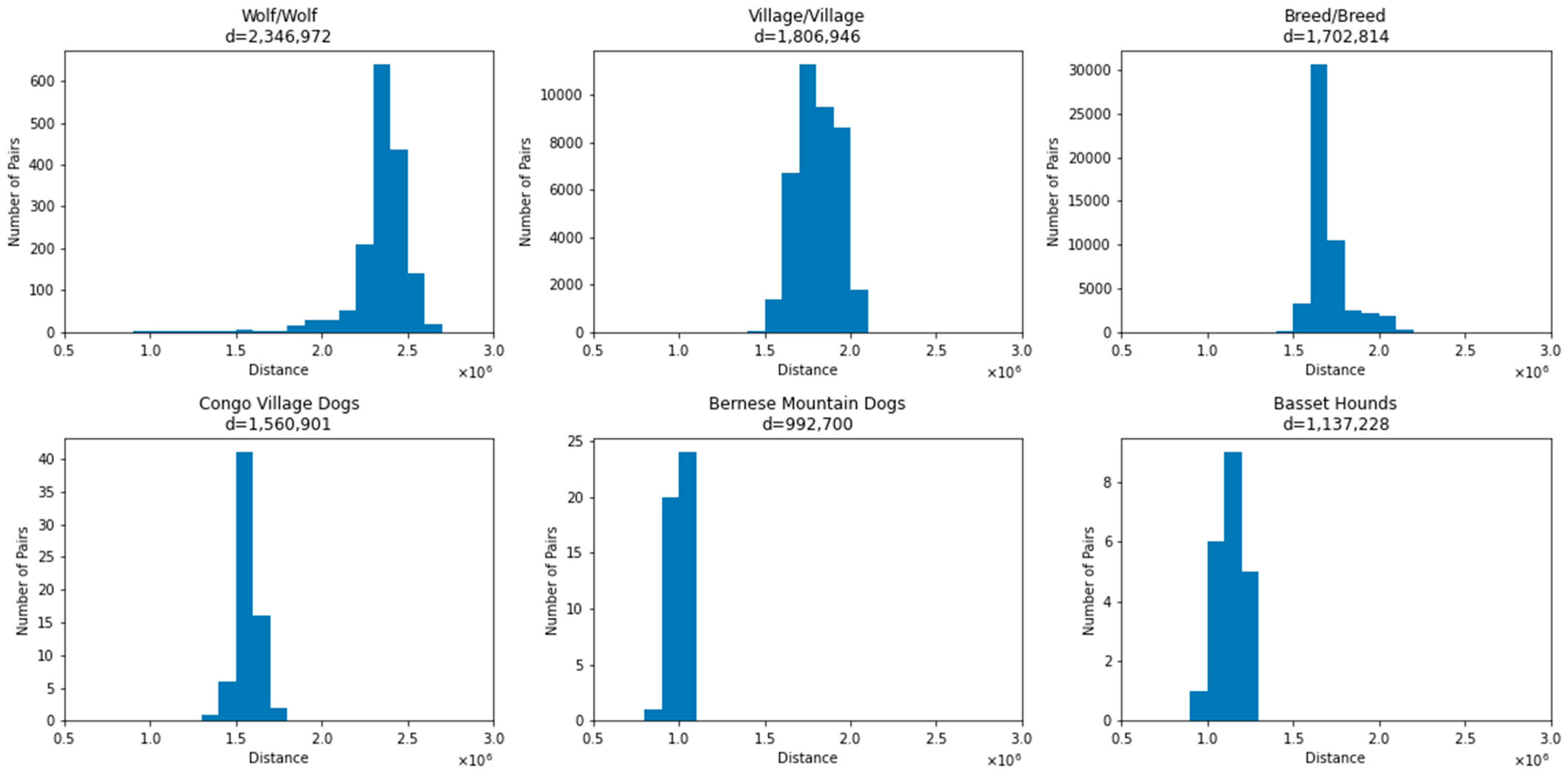
3.1. Average Number of Differences between Canines
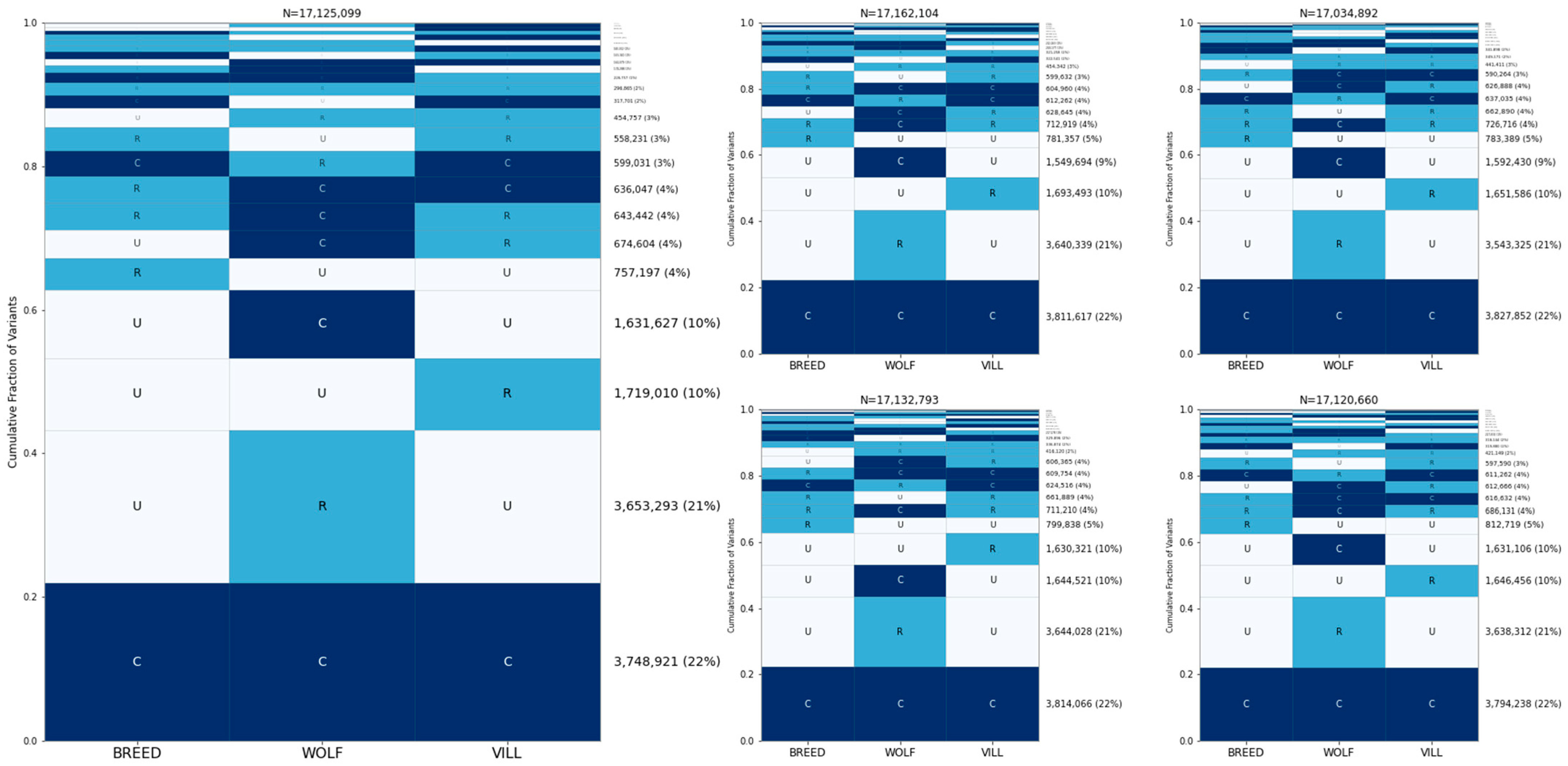
3.2. Sharing of Alleles among Breed Dogs, Wolves, and Village Dogs
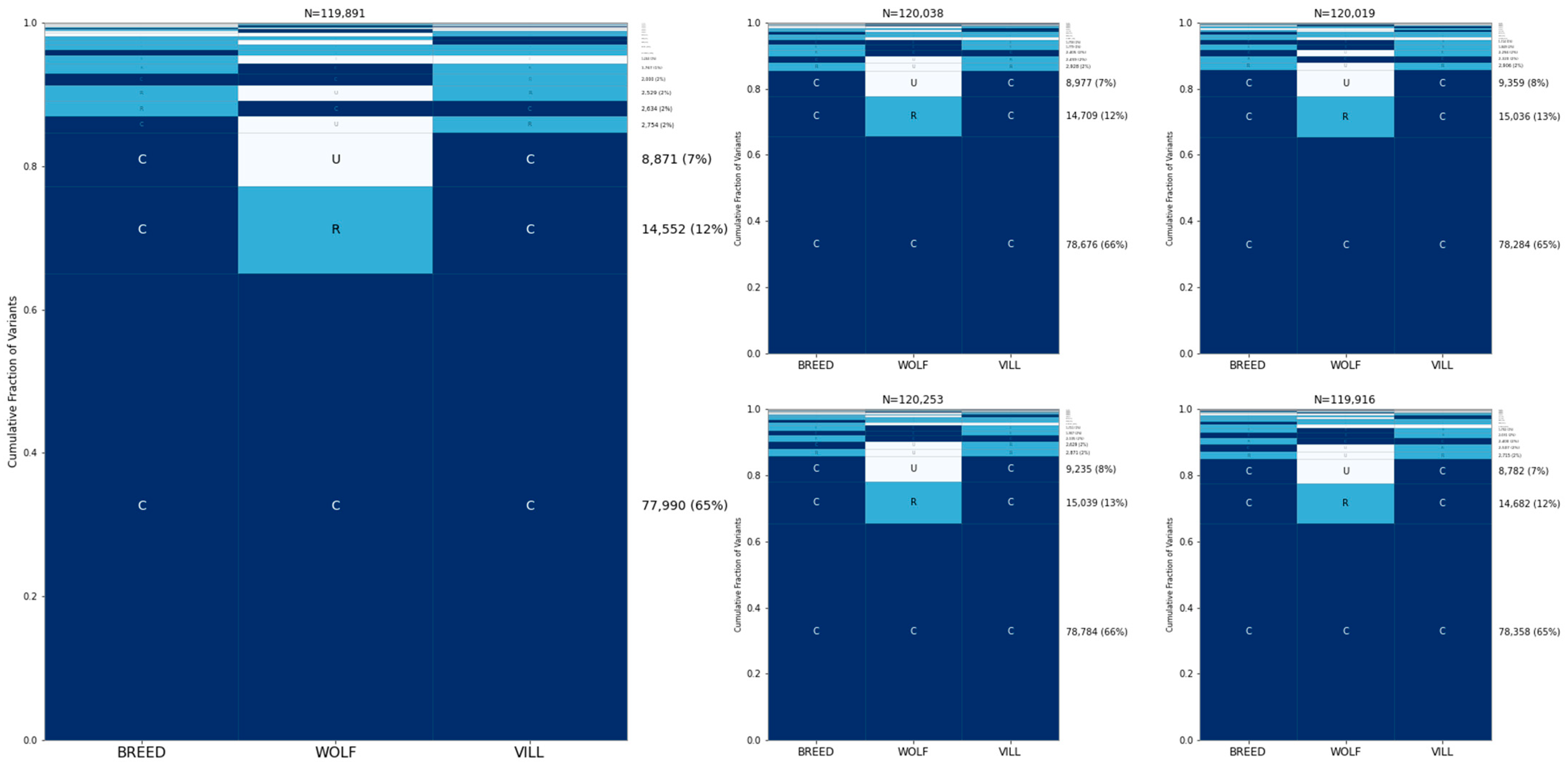
3.3. Allele Sharing within Village Dogs and Breed Clades
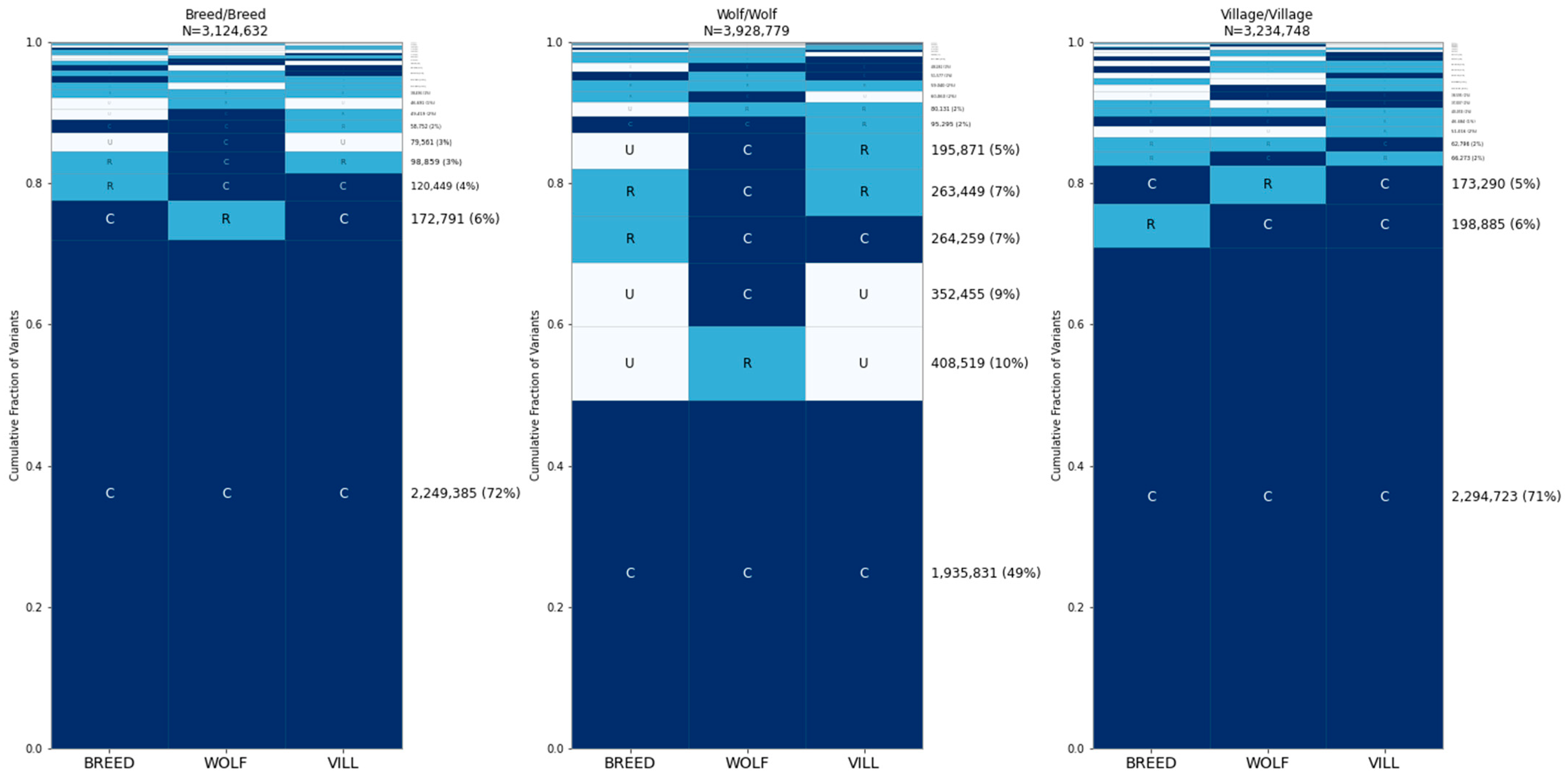
3.4. SNP Microarrays Are Skewed toward Globally Common Sites
3.5. Common Variants Account for Most of the Differences between Individual Dogs and Wolves
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skoglund, P.; Ersmark, E.; Palkopoulou, E.; Dalen, L. Ancient wolf genome reveals an early divergence of domestic dog ancestors and admixture into high-latitude breeds. Curr. Biol. 2015, 25, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Botigue, L.R.; Song, S.; Scheu, A.; Gopalan, S.; Pendleton, A.L.; Oetjens, M.; Taravella, A.M.; Seregely, T.; Zeeb-Lanz, A.; Arbogast, R.M.; et al. Ancient European dog genomes reveal continuity since the Early Neolithic. Nat. Commun. 2017, 8, 16082. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, A.; Frantz, L.; Schmidt, R.; Ersmark, E.; Lebrasseur, O.; Girdland-Flink, L.; Lin, A.T.; Stora, J.; Sjogren, K.G.; Anthony, D.; et al. Origins and genetic legacy of prehistoric dogs. Science 2020, 370, 557–564. [Google Scholar] [CrossRef]
- Frantz, L.A.; Mullin, V.E.; Pionnier-Capitan, M.; Lebrasseur, O.; Ollivier, M.; Perri, A.; Linderholm, A.; Mattiangeli, V.; Teasdale, M.D.; Dimopoulos, E.A.; et al. Genomic and archaeological evidence suggest a dual origin of domestic dogs. Science 2016, 352, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Freedman, A.H.; Gronau, I.; Schweizer, R.M.; Ortega-Del Vecchyo, D.; Han, E.; Silva, P.M.; Galaverni, M.; Fan, Z.; Marx, P.; Lorente-Galdos, B.; et al. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 2014, 10, e1004016. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D.; Ortega-Del Vecchyo, D.; O‘Brien, D.P.; Taylor, J.F.; Ramirez, O.; Vila, C.; Marques-Bonet, T.; Schnabel, R.D.; Wayne, R.K.; Lohmueller, K.E. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc. Natl. Acad. Sci. USA 2016, 113, 152–157. [Google Scholar] [CrossRef]
- Parker, H.G.; Shearin, A.L.; Ostrander, E.A. Man’s best friend becomes biology’s best in show: Genome analyses in the domestic dog. Annu. Rev. Genet. 2010, 44, 309–336. [Google Scholar] [CrossRef]
- Shearin, A.L.; Ostrander, E.A. Leading the way: Canine models of genomics and disease. Dis. Model. Mech. 2010, 3, 27–34. [Google Scholar] [CrossRef]
- Axelsson, E.; Ljungvall, I.; Bhoumik, P.; Conn, L.B.; Muren, E.; Ohlsson, A.; Olsen, L.H.; Engdahl, K.; Hagman, R.; Hanson, J.; et al. The genetic consequences of dog breed formation-Accumulation of deleterious genetic variation and fixation of mutations associated with myxomatous mitral valve disease in cavalier King Charles spaniels. PLoS Genet. 2021, 17, e1009726. [Google Scholar] [CrossRef]
- Mooney, J.A.; Yohannes, A.; Lohmueller, K.E. The impact of identity by descent on fitness and disease in dogs. Proc. Natl. Acad. Sci. USA 2021, 118, e2019116118. [Google Scholar] [CrossRef]
- Bannasch, D.; Famula, T.; Donner, J.; Anderson, H.; Honkanen, L.; Batcher, K.; Safra, N.; Thomasy, S.; Rebhun, R. The effect of inbreeding, body size and morphology on health in dog breeds. Canine Med. Genet. 2021, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Yordy, J.; Kraus, C.; Hayward, J.J.; White, M.E.; Shannon, L.M.; Creevy, K.E.; Promislow, D.E.L.; Boyko, A.R. Body size, inbreeding, and lifespan in domestic dogs. Conserv. Genet. 2020, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Shannon, L.M.; Boyko, R.H.; Castelhano, M.; Corey, E.; Hayward, J.J.; McLean, C.; White, M.E.; Abi Said, M.; Anita, B.A.; Bondjengo, N.I.; et al. Genetic structure in village dogs reveals a Central Asian domestication origin. Proc. Natl. Acad. Sci. USA 2015, 112, 13639–13644. [Google Scholar] [CrossRef] [PubMed]
- Boyko, A.R. The domestic dog: Man’s best friend in the genomic era. Genome Biol. 2011, 12, 216. [Google Scholar] [CrossRef]
- Boyko, A.R.; Boyko, R.H.; Boyko, C.M.; Parker, H.G.; Castelhano, M.; Corey, L.; Degenhardt, J.D.; Auton, A.; Hedimbi, M.; Kityo, R.; et al. Complex population structure in African village dogs and its implications for inferring dog domestication history. Proc. Natl. Acad. Sci. USA 2009, 106, 13903–13908. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., 3rd; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Hoeppner, M.P.; Lundquist, A.; Pirun, M.; Meadows, J.R.; Zamani, N.; Johnson, J.; Sundstrom, G.; Cook, A.; FitzGerald, M.G.; Swofford, R.; et al. An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PLoS ONE 2014, 9, e91172. [Google Scholar] [CrossRef]
- Hayward, J.J.; Castelhano, M.G.; Oliveira, K.C.; Corey, E.; Balkman, C.; Baxter, T.L.; Casal, M.L.; Center, S.A.; Fang, M.; Garrison, S.J.; et al. Complex disease and phenotype mapping in the domestic dog. Nat. Commun. 2016, 7, 10460. [Google Scholar] [CrossRef]
- Plassais, J.; Kim, J.; Davis, B.W.; Karyadi, D.M.; Hogan, A.N.; Harris, A.C.; Decker, B.; Parker, H.G.; Ostrander, E.A. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat. Commun. 2019, 10, 1489. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drogemuller, C.; Leeb, T.; Dog Biomedical Variant Database, C. A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef]
- Meadows, J.R.S.; Kidd, J.M.; Wang, G.D.; Parker, H.G.; Schall, P.Z.; Bianchi, M.; Christmas, M.J.; Bougiouri, K.; Buckley, R.M.; Hitte, C.; et al. Genome sequencing of 2000 canids by the Dog10K consortium advances the understanding of demography, genome function and architecture. Genome Biol 2023, 24, 187. [Google Scholar] [CrossRef]
- Wang, C.; Wallerman, O.; Arendt, M.L.; Sundstrom, E.; Karlsson, A.; Nordin, J.; Makelainen, S.; Pielberg, G.R.; Hanson, J.; Ohlsson, A.; et al. A novel canine reference genome resolves genomic architecture and uncovers transcript complexity. Commun. Biol. 2021, 4, 185. [Google Scholar] [CrossRef]
- Jagannathan, V.; Hitte, C.; Kidd, J.M.; Masterson, P.; Murphy, T.D.; Emery, S.; Davis, B.; Buckley, R.M.; Liu, Y.H.; Zhang, X.Q.; et al. Dog10K_Boxer_Tasha_1.0: A Long-Read Assembly of the Dog Reference Genome. Genes 2021, 12, 847. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.J.; Field, M.A.; Ferguson, J.M.; Dudchenko, O.; Keilwagen, J.; Rosen, B.D.; Johnson, G.S.; Rice, E.S.; Hillier, D.; Hammond, J.M.; et al. Chromosome-length genome assembly and structural variations of the primal Basenji dog (Canis lupus familiaris) genome. BMC Genom. 2021, 22, 188. [Google Scholar] [CrossRef]
- Player, R.A.; Forsyth, E.R.; Verratti, K.J.; Mohr, D.W.; Scott, A.F.; Bradburne, C.E. A novel canis lupus familiaris reference genome improves variant resolution for use in breed-specific GWAS. Life Sci. Alliance 2021, 4. [Google Scholar] [CrossRef]
- Sinding, M.S.; Gopalakrishnan, S.; Raundrup, K.; Dalen, L.; Threlfall, J.; Darwin Tree of Life Barcoding Collective; Wellcome Sanger Institute Tree of Life Programme; Wellcome Sanger Institute Scientific Operations: DNA Pipelines Collective; Tree of Life Core Informatics Collective; Darwin Tree of Life Consortium; et al. The genome sequence of the grey wolf, Canis lupus Linnaeus 1758. Wellcome Open Res. 2021, 6, 310. [Google Scholar] [CrossRef] [PubMed]
- Halo, J.V.; Pendleton, A.L.; Shen, F.; Doucet, A.J.; Derrien, T.; Hitte, C.; Kirby, L.E.; Myers, B.; Sliwerska, E.; Emery, S.; et al. Long-read assembly of a Great Dane genome highlights the contribution of GC-rich sequence and mobile elements to canine genomes. Proc. Natl. Acad. Sci. USA 2021, 118, e2016274118. [Google Scholar] [CrossRef]
- Schall, P.Z.; Winkler, P.A.; Petersen-Jones, S.M.; Yuzbasiyan-Gurkan, V.; Kidd, J.M. Genome-wide methylation patterns from canine nanopore assemblies. G3 Genes Genomes Genet. 2023, 13, jkad203. [Google Scholar] [CrossRef]
- Bredemeyer, K.R.; vonHoldt, B.M.; Foley, N.M.; Childers, I.R.; Brzeski, K.E.; Murphy, W.J. The value of hybrid genomes: Building two highly contiguous reference genome assemblies to advance Canis genomic studies. J. Hered. 2024, 115, 480–486. [Google Scholar] [CrossRef]
- Clark, A.G.; Hubisz, M.J.; Bustamante, C.D.; Williamson, S.H.; Nielsen, R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005, 15, 1496–1502. [Google Scholar] [CrossRef]
- Lachance, J.; Tishkoff, S.A. SNP ascertainment bias in population genetic analyses: Why it is important, and how to correct it. Bioessays 2013, 35, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Biddanda, A.; Rice, D.P.; Novembre, J. A variant-centric perspective on geographic patterns of human allele frequency variation. Elife 2020, 9, e60107. [Google Scholar] [CrossRef] [PubMed]
- Gronau, I.; Hubisz, M.J.; Gulko, B.; Danko, C.G.; Siepel, A. Bayesian inference of ancient human demography from individual genome sequences. Nat. Genet. 2011, 43, 1031–1034. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Byrska-Bishop, M.; Evani, U.S.; Zhao, X.; Basile, A.O.; Abel, H.J.; Regier, A.A.; Corvelo, A.; Clarke, W.E.; Musunuri, R.; Nagulapalli, K.; et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 2022, 185, 3426–3440.e19. [Google Scholar] [CrossRef]
- Pfahler, S.; Distl, O. Effective population size, extended linkage disequilibrium and signatures of selection in the rare dog breed lundehund. PLoS ONE 2015, 10, e0122680. [Google Scholar] [CrossRef] [PubMed]
- Kettunen, A.; Daverdin, M.; Helfjord, T.; Berg, P. Cross-Breeding Is Inevitable to Conserve the Highly Inbred Population of Puffin Hunter: The Norwegian Lundehund. PLoS ONE 2017, 12, e0170039. [Google Scholar] [CrossRef]
- Fan, Z.; Silva, P.; Gronau, I.; Wang, S.; Armero, A.S.; Schweizer, R.M.; Ramirez, O.; Pollinger, J.; Galaverni, M.; Ortega Del-Vecchyo, D.; et al. Worldwide patterns of genomic variation and admixture in gray wolves. Genome Res. 2016, 26, 163–173. [Google Scholar] [CrossRef]
- Nguyen, A.K.; Schall, P.Z.; Kidd, J.M. A map of canine sequence variation relative to a Greenland wolf outgroup. Mamm. Genome 2024. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clade | Number of Samples | Clade | Number of Samples |
|---|---|---|---|
| Alpine | 67 | Mastiff | 97 |
| American Terriers | 31 | Pointer | 138 |
| Asian | 135 | Retriever | 36 |
| Belgian Herders | 43 | Scenthound | 222 |
| Continental Herders | 38 | Scottish Terrier | 40 |
| English Terriers | 53 | Spaniel | 62 |
| Flockguard Sighthound | 145 | Spitz | 152 |
| German Shepherd | 32 | UK Herding | 46 |
| Hungary | 43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funk, M.W.; Kidd, J.M. A Variant-Centric Analysis of Allele Sharing in Dogs and Wolves. Genes 2024, 15, 1168. https://doi.org/10.3390/genes15091168
Funk MW, Kidd JM. A Variant-Centric Analysis of Allele Sharing in Dogs and Wolves. Genes. 2024; 15(9):1168. https://doi.org/10.3390/genes15091168
Chicago/Turabian StyleFunk, Matthew W., and Jeffrey M. Kidd. 2024. "A Variant-Centric Analysis of Allele Sharing in Dogs and Wolves" Genes 15, no. 9: 1168. https://doi.org/10.3390/genes15091168