Abstract
The mitochondrial genome (mitogenome) Rhagastis binoculata (Matsumura, 1909), an endemic moth species in Taiwan, was sequenced and analyzed. The complete circular mitogenome of R. binoculata is 15,303 bp and contains 13 protein-coding genes, 22 transfer RNA genes, 2 ribosomal RNA genes, and an AT-rich control region. The mitogenome has an overall nucleotide composition of 41.2% A, 11.9% C, 7.5% G, and 39.4% T, with an AT content of 80.6%. Of the protein-coding genes (PCGs), 12 start with ATG, ATT, and ATC, and COX1 starts with a “CGA” codon. All of the stop codons are “TAA, TAG, or T”. Our phylogenetic analysis of 21 species of Sphingidae insects suggests that R. binoculata is clustered with Rhagastis mongoliana, which belongs to the subfamily Macroglossinae.
1. Introduction
Lepidoptera is a large insect order with more than 160,000 species [1], including butterflies and moths. Many Lepidoptera species serve as important models in ecology and evolutionary biology [2]. In terms of lepidopteran insects, the mitochondrial genomes (mitogenomes) are characterized by a control region that is rich in adenine and thymine (A + T) and typically comprises 37 genes, including 13 conserved protein-coding genes (PCGs), 22 tRNAs, 2 rRNAs, and a non-coding region [3,4,5]. The size of the mitogenomes also varies from 15,000 to over 16,000 bp [3,4]. This variation in the mitogenomes is primarily due to differences in the lengths of non-coding regions, especially the control region [6]. Based on these characteristics, the mitogenomes of species in the order Lepidoptera are extensively used for research in population genetics, phylogeography, phylogenetics, and molecular taxonomy [7,8,9]. The mitogenome, in particular, is an excellent resource for phylogenetic analysis owing to its uncomplicated structure, maternal mode of inheritance, limited recombination, and significant evolutionary conservation [3,4].
The family Sphingidae, commonly known as hawk moths or sphinx moths, comprises a significant number of genera and species. There are approximately 200 genera and around 1350 species within this family [10], which is distributed globally, except in extreme environments like Antarctica and certain deserts [11]. Sphingidae moths play a crucial role in both ecological and economic contexts. Ecologically, they are important pollinators and serve as indicators of environmental health.
Rhagastis binoculata (Matsumura, 1909) is one species belonging to the family Sphingidae. It is an endemic moth species in Taiwan. According to recorded data, the R. binoculata species is distributed throughout low (ca. 161 m) to medium (ca. 2350 m) altitudes of mountain areas in Taiwan [12]. The males are easily drawn to light, whereas females are less frequently captured using light traps [13]. The wing length of R. binoculata ranges from 54 to 70 mm. R. binoculata adult males can be identified by a pinkish-gray oval patch that spans the postmedian lines and a more prominent black spot on the upper side of the forewing [13]. The underside of both the forewing and hindwing is generally orange in color, with patterns that are more diffused compared to those seen in R. dichroae, yet still discernible [13]. In contrast, the female shares similar traits with the male but features broader wings and a slightly lighter ground color. Additionally, the grayish zigzag medial line on the underside of both the forewing and hindwing is more noticeable in females [13]. The male genitalia of R. binoculata bear resemblance to those of R. everetti, though with some distinctive differences. The uncus and gnathos are comparatively shorter and thicker [13]. The valva is rounded, with the basal part narrower than the terminal part. The sacculus is very short, slightly curved, and ends in a blunt apex [13]. These anatomical details are essential for precise species identification and understanding the phylogenetic relationships within the genus Rhagastis.
The major host plants of R. binoculata larvae are the Chinese hydrangea (Hydrangea chinensis) and narrow-petaled hydrangea (H. angustipetala) [14]. Therefore, there is no record of agricultural losses in this species. The morphology of the R. binoculata larva consists of a green body color with distinctive white markings resembling clouds on its lateral sides. Positioned atop the hump behind the head are a pair of eyespots. Furthermore, a prominent central blue line runs along the dorsal part of the larval body. In this study, R. binoculata larvae displaying nucleopolyhedrosis were sampled from the Chinese hydrangea at Tianshang Mountain Trail, New Taipei City, Taiwan (24°57′26.0″ N 121°26′39.4″ E), at low altitude (ca. 400 m).
Nucleopolyhedrosis is a symptom caused by nucleopolyhedrovirus (NPV) infection. The NPV belongs to the genus Alphabaculovirus in the family Baculoviridae [15]. Based on our knowledge, baculoviruses are insect-specific pathogens, and over 600 insect host species have been reported for baculoviral infection [16]. Furthermore, since the 1980s, recombinant baculoviruses have been established as baculovirus expression vector systems (BEVSs) and widely applied to foreign protein expression in insects [16]. To date, the most commonly used commercial BEVSs are based on the Autographa californica multiple nucleopolyhedrovirus (AcMNPV) systems; however, the large-scale production of recombinant protein is still under development [17]. Baculoviruses usually exhibit a narrow host range [18]. In terms of host specificity, the identification of the insect species is merely important for the discovery of a new NPV infection in the field. Moreover, due to the large body size of R. binoculata larvae, it is considered to have the potential to be developed as a BEVS with highly foreign protein productivity. To explore this potential and to further understand the host of this NPV, the mitogenome of an R. binoculata larva was decoded with next-generation sequencing (NGS). These data could provide more information on the Macroglossinae subfamily and contribute to the study of the insect virus’s host range.
2. Materials and Methods
2.1. Sample Information, Collections, and DNA Extraction
In this study, a diseased R. binoculata larva, which showed nucleopolyhedrosis, was collected from the H. chinensis at Tianshang Mountain Trail, New Taipei City, Taiwan (24°57′26.0″ N 121°26′39.4″ E), at an altitude of ca. 400 m. The diseased-larva sample was deposited at the Insect Pathology and Genomics Laboratory (IPL) at the Department of Entomology, National Chung Hsing University, under voucher number NCHU_IPL_41. Total genomic DNA was extracted from the diseased R. binoculata larva using a genomic DNA extraction Kit (Geneaid Biotech Ltd., New Taipei City, Taiwan) following the user manual.
2.2. Genomic Sequencing and Data Analysis
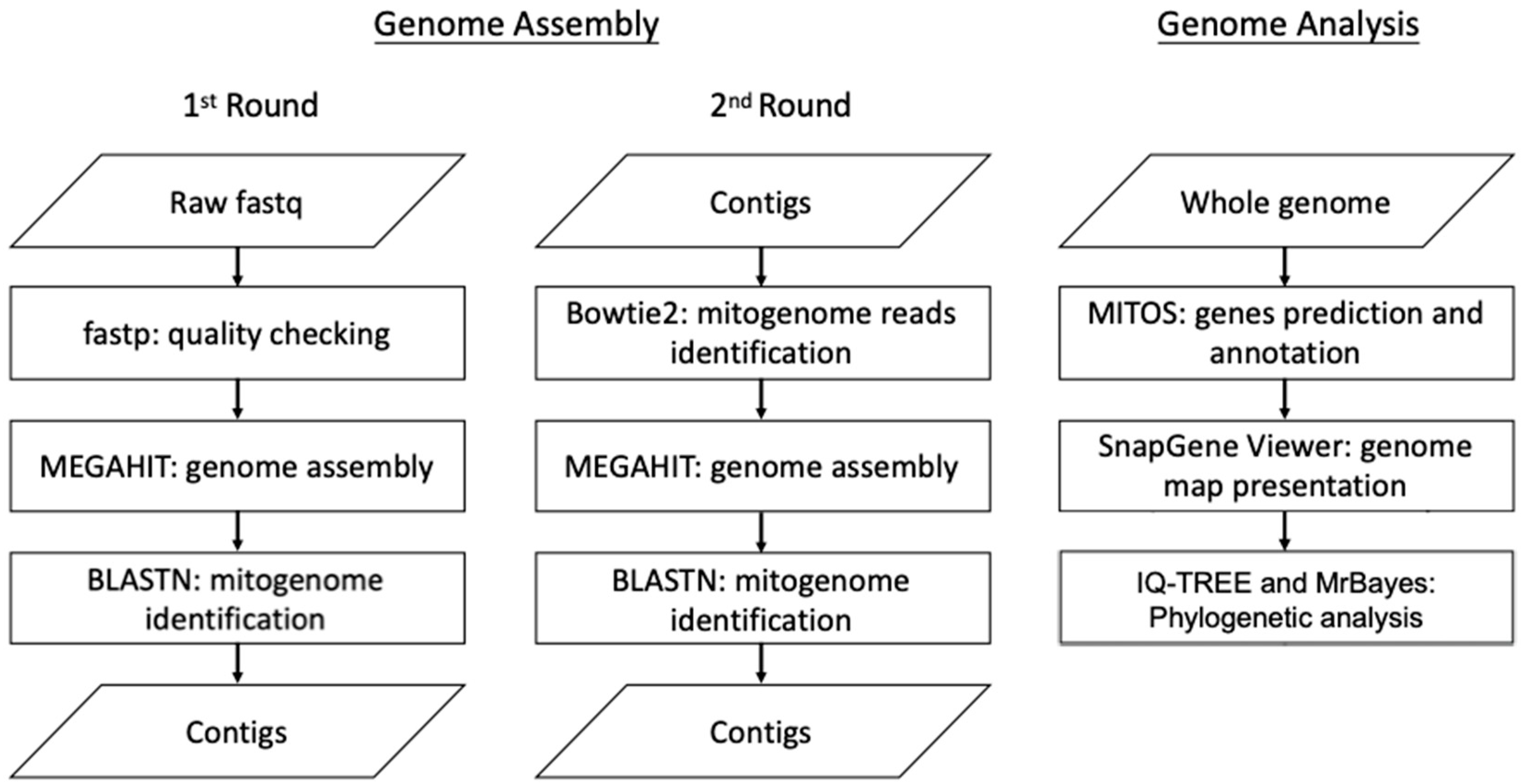
DNA sequencing was performed with an Illumina NovaSeq 6000 sequencer (Illumina, San Diego, CA, USA) with paired-end (PE) technology for 2 × 150 bp. The total PE reads were conducted for sequencing adapter identification and then trimmed with cutadapt [19]. Ambiguous bases and bases with lower quality values were removed with PRINseq [20] from either the 5′- or 3′ end. The quality check of the trimmed reads was performed using fastp [21]. These trimmed reads were then subjected to genome assembly and analysis. This workflow is summarized in Figure 1. The genome assembly was performed with a MEGAHIT de novo assembler with quality PE reads, as in a previous study [22]. Briefly, the quality reads were subjected to a 1st round of genome assembly by the MEGAHIT de novo assembler. The assembled contigs were then checked with NCBI BLASTn to identify the contigs related to the mitogenome sequences. All the quality reads were mapped to the 1st assembled mitogenome-related contigs, and the mapped reads were subjected to a 2nd round of genome assembly with the MEGAHIT de novo assembler to obtain the final genome draft. In total, four contigs were obtained, and their orientation was determined by aligning them against the reference mitogenome of Clanis bilineata (GenBank: MK804156.1). The draft mitogenome was then subjected to gap filling using PCR with four primer sets (Supplementary Table S1) to complete the circular mitogenome. The mitogenome of R. binoculata was annotated using MITOS WebServer (http://mitos.bioinf.uni-leipzig.de/index.py, accessed on 23 March 2023) [23], and the start and stop codons of the protein-coding genes (PCGs) were checked manually, in accordance with the study of Cameron et al., 2014 [24]. The genome map was presented by SnapGene Viewer (www.snapgene.com).
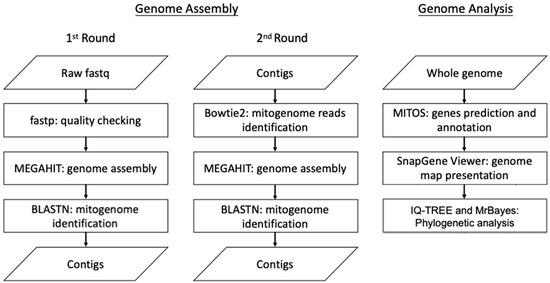
Figure 1.
The workflow of genome assembly and analysis.
2.3. Species-Level Confirmation of Mitogenome
To confirm that the mitogenome from the diseased R. binoculata larva (Figure 2a) was identical to that of the R. binoculata adult, an adult R. binoculata specimen was obtained from the National Museum of Natural Science (NMNS) under voucher number NCHU_IPL_41-14 (Figure 2b). DNA samples were extracted from both specimens and subjected to PCR amplification with a COX1 primer set (Supplementary Table S1). The PCR products were then sequenced using Sanger sequencing, and the sequences were aligned with MAFFT (v7.551) (https://mafft.cbrc.jp/alignment/server/, accessed on 27 August 2024) to confirm the species of the diseased larva.

Figure 2.
The larva and adult of R. binoculata (Matsumura, 1909). (a) The diseased larva (NCHU_IPL_41) with the symptom of nucleopolyhedrosis. (b) The adult specimen of R. binoculata (Matsumura, 1909) (NCHU_IPL_41-14) from the National Museum of Natural Science (NMNS).
2.4. Phylogenetic Analyses
To better understand the classification of R. binoculata, a phylogenetic analysis of 21 moth species in Sphingidae was performed based on whole mitogenomes (Supplementary Table S2) [5,25,26,27,28,29]. To select the species of Sphingidae, the COX1 gene was used to search the closely related species with NCBI BlastN [30], and species with a similarity between 90% and 100% were selected. The mitogenome of C. bilineata was also included in the phylogenetic analysis because the NPV found in R. binoculata is the most closely related to the NPV from C. bilineata. The control regions were trimmed before alignment using MUSCLE (v5.1) [31]. After alignment, the tree model was evaluated with ModelFinder [32,33], and then, the phylogenetic tree was constructed using IQ-TREE [34] with the model GTR + F + I + G4 and MrBayes 3.2.6 [35,36,37] with the model GTR + I + γ.
3. Results
3.1. Sequencing Summary, Mitogenome Assembly, and Annotation
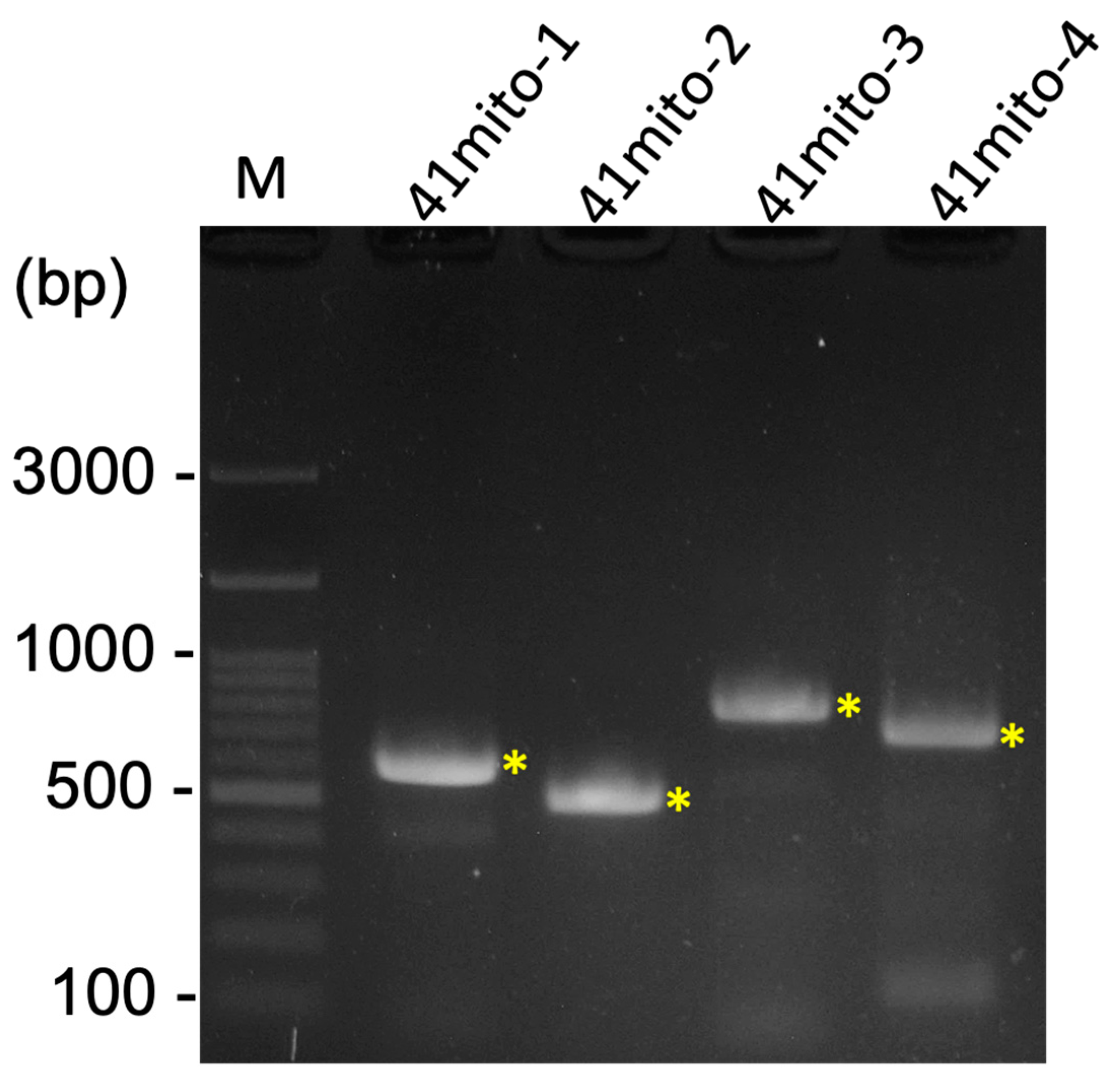
A total of 62,429,098 clean raw reads were obtained through NGS, and the raw sequencing data were deposited in the NCBI database with the Sequence Read Archive (SRA) accession number PRJNA1045710. Of these reads, 59,799 reads belonged to the mitogenome of R. binoculata. After genomic assembly, four contigs were obtained, and the mitogenome was then concatenated and completed using PCR (Figure 3). The final completed mitogenome of R. binoculata was 15,303 bp in length with a 100% coverage and 107-fold mean depth (Supplementary Figure S1). The nucleotide composition was 41.2% A, 11.9% C, 7.5% G, and 39.4% T, with an A+T bias of 80.6% (Figure 4). In terms of the species-level confirmation, the COX1 sequence of the diseased larva and the adult specimen of R. binoculata showed a 100% similarity in identity, indicating that these samples were the same species (Figure 5).
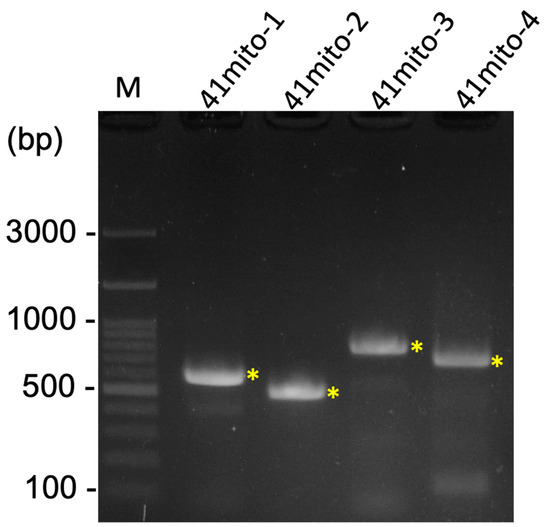
Figure 3.
The result of four PCRs for the gap closing of the R. binoculata mitogenome. M = 100 bp DNA marker; bp = base pair; “*” indicates the target amplified PCR bands.
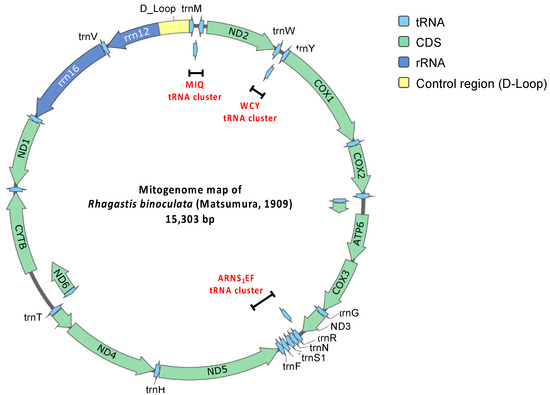
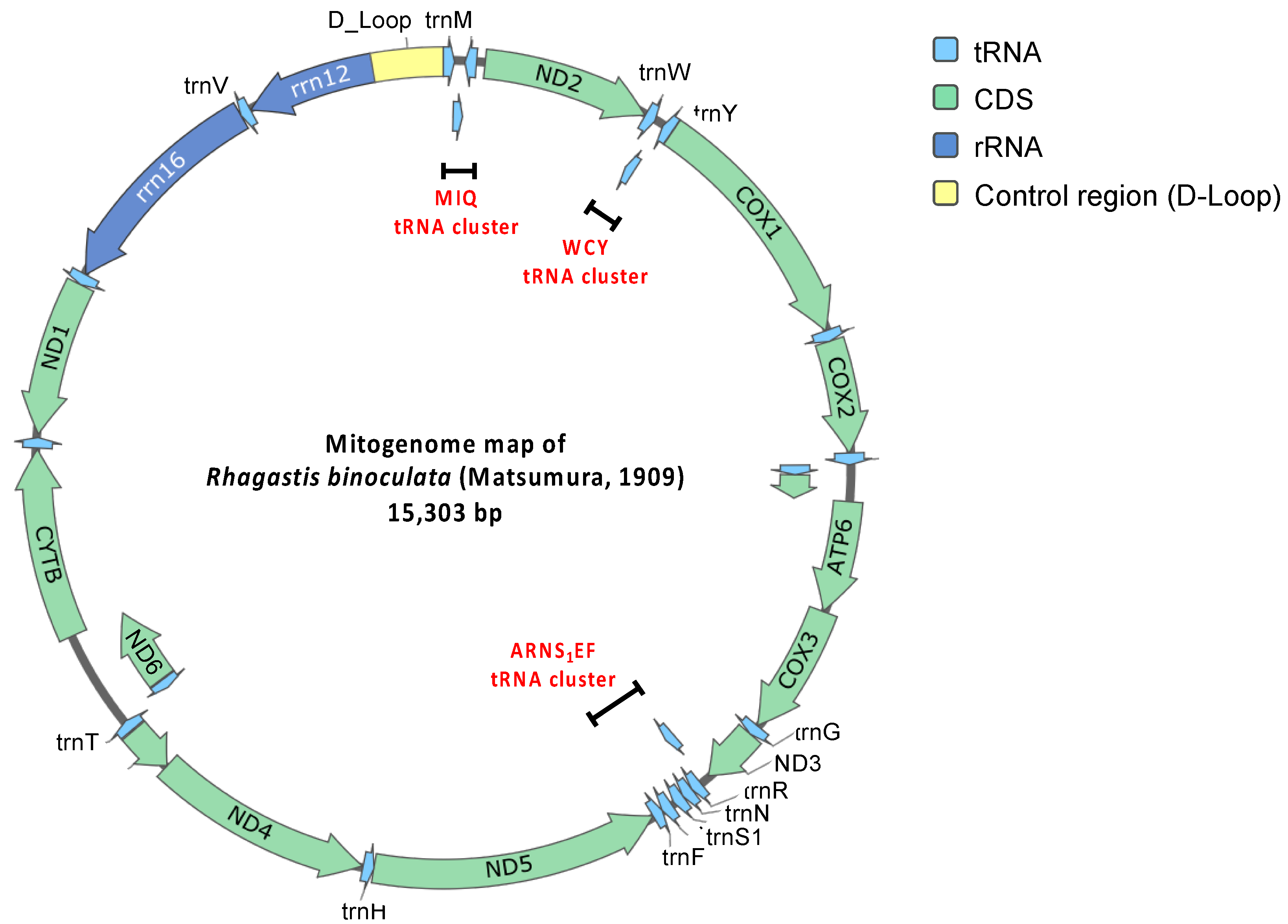
Figure 4.
Circular mitogenome map of R. binoculata (Matsumura, 1909).

Figure 5.
The confirmation of the identity of the diseased larva (NCHU_IPL_41) and adult R. binoculata specimen (NCHU_IPL_41-14) using the COXI gene PCR. (a,b), Sanger sequencing. (c). M = 100 bp DNA marker; bp = base pair; “*” indicates the target amplified PCR bands. The alignment of the sequences was performed by MAFFT (v7.551) https://mafft.cbrc.jp/alignment/server/, accessed on 27 August 2024.
3.2. Protein-Coding Genes
The mitogenome consists of 13 protein-coding genes, 22 transfer RNA (tRNA) genes, 2 ribosomal RNA (rRNA) genes, and a major non-coding AT-rich control region (Table 1; Figure 4). Based on the comparison of 13 protein-coding genes, several start and stop codons of R. binoculata are different to those of R. mongoliana. For ND2 (NADH dehydrogenase 2), the start codon (ATC) in the mitogenome of R. binoculata is different from that of R. mongoliana, which is ATT (start codon). For COX1, COX2, and ND5 (NADH dehydrogenase 5), the stop codons (T) are different from those of R. mongoliana (ATT, CTT, and TTT, respectively). For ATP8 (ATP synthase of subunit 8) and ND5, the start codons are ATT in R. binoculata, while the start codons found in R. mongoliana are ATC. The stop codon (TCT) of ND5 in R. binoculata is different from that of R. mongoliana (stop codon = TTT). Moreover, the stop codon of ND1 in R. binoculata is also different from that of R. mongoliana, which is TAA. These differences in codons usage might imply translational efficacy or RNA stability in different insect species.

Table 1.
The mitochondrial genes of the Rhagastis binoculata mitogenome.
3.3. Transfer RNA and Ribosomal RNA Genes
3.4. Phylogenetic Analysis
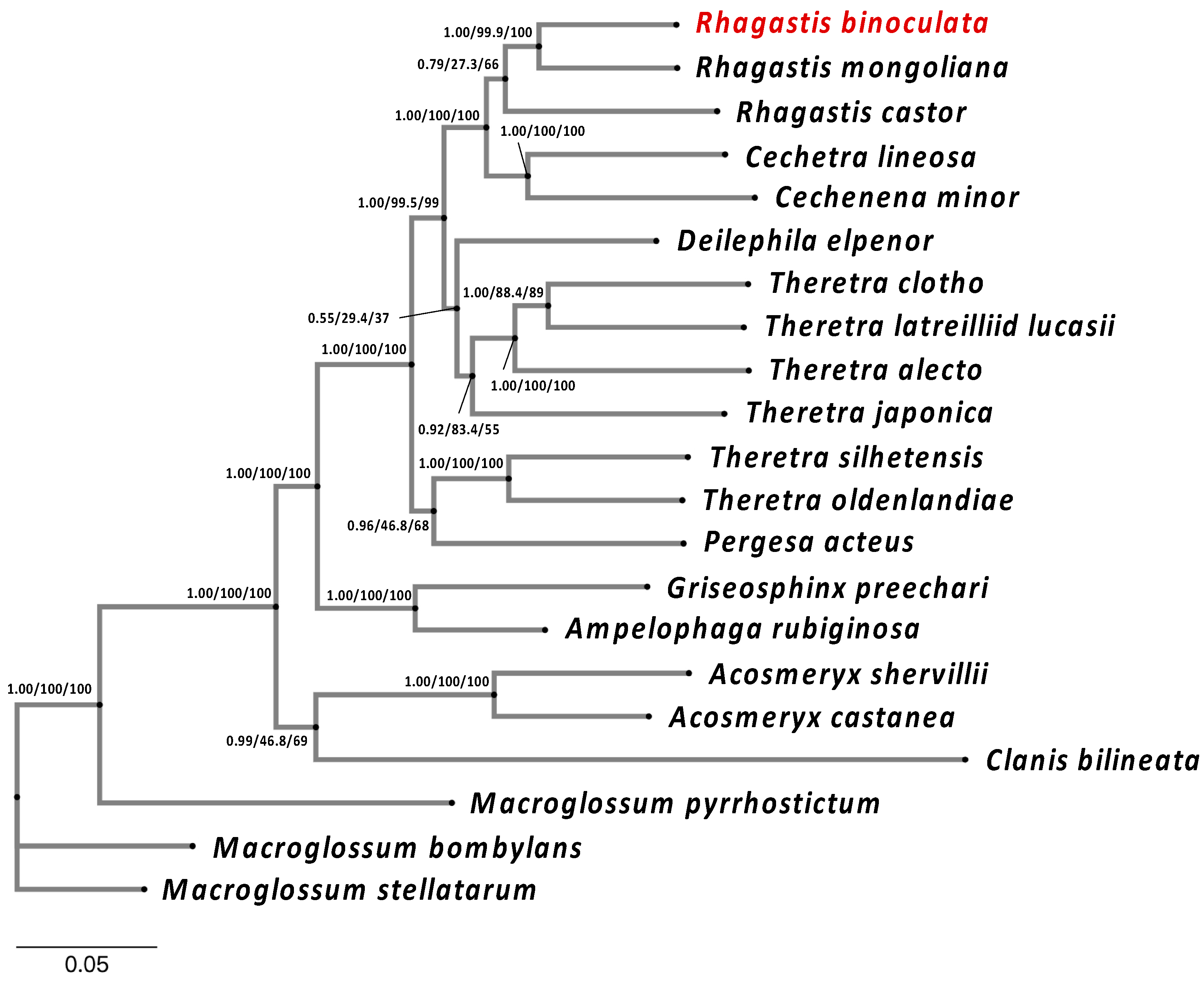
Both the MrBayes and IQ-TREE results indicate that R. mongoliana (GenBank: OL622048.1) is the closest species to R. binoculata (Figure 6) and that they both belong to the subfamily Macroglossinae. The phylogenetic trees from these two phylogenetic analyses had identical structures, which suggests that the results of the phylogenetic analysis of R. binoculata are reliable.
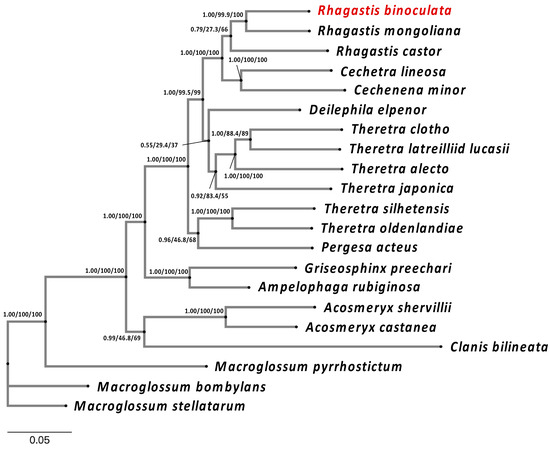
Figure 6.
The phylogenetic analysis of R. binoculata (Matsumura, 1909) showing its relation to other close species. The phylogenetic analysis is based on whole mitogenomes, except for the control regions (A+T-rich) of 21 species in Sphingidae. The phylogenetic tree was constructed using both MrBayes and IQ-TREE, applying the Bayesian inference (BI) and maximum-likelihood (ML) methods. Branch support values are represented as Posterior Probability (PP) (for BI), UFBoot2 (for ML), and SH-Alrt (for ML) (PP/UFBoot2/SH-Alrt).
4. Discussion
After genomic sequencing, assembly, analysis, and PCR validation, the size of R. binoculata’s mitogenome is 15,303 bp. This mitogenome size is within the range of those of other Lepidopteran insects, which generally range from 15 to 16 kb [38]. The A+T content of insects varies, ranging from 64% (termites) to 86.7% (bees) [24]. Within the Lepidopteran species, the mitogenomes are A+T-rich (average% = 80.49% ± 0.95%), and also ranging from 76.01% (Acraea polis, Nymphalidae) to 83.13% (Hesperia comma, Hesperiidae) [39]. The A+T bias of R. binoculata mitogenome is 80.6%, aligning with the A+T richness typical of Lepidopteran species. All mitogenomes in Lepidoptera contain 37 genes and an AT-rich region, including 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, and 2 ribosomal RNA (rRNA) genes. In R. binoculata, 23 genes are located on the forward strand, including 9 PCGs and 14 tRNA genes, while the remaining 14 genes are on the reverse strand. These characteristics have also been observed in previous research [38].
In typical Lepidopteran mitogenomes, the start codon of COX1 is CGA, while the other 12 PCGs have ATN as their start codon [40]. However, there are some exceptions. The supposed start codon in COX1 should be replaced by ATA or ATT in five primitive clades (superfamilies Adeloidea, Nepticuloidea and Hepialoidea) and some of Ditrysia (the superfamilies Tineoidea and Zygaenoidea). Additionally, the start codon “GTG” is partially found in the COX2 and ND1 genes of the superfamily Zygaenoidea [38] and is also present in other species’ mitogenomes [41]. In contrast to the diversity of start codons, there are only four types of stop codons: TAA, TAG, TA, and T [38]. TAA is the most common stop codon in PCGs. COX1 and COX2 use the incomplete codon T as stop codons, while the stop codon of ND5 varies. These conserved mitogenomic features are common in invertebrates [42,43,44], and these non-standard stop codons (T and TA) can still be recognized by endonucleases during the transcription of polycistronic pre-mRNA, resulting in a functional stop codon through the polyadenylation of contiguous PCGs [45,46]. In this study of the start codons of PCGs in R. binoculata, the start codon of COX1 is CGA, while the start codons of the other 12 codons are ATB (B=C or G or T). Among the stop codons, COX1, COX2, and ND5 have incomplete stop codons (T).
There are 22 tRNA genes in the mitogenomes of R. binoculata. Fourteen of them are forward strand, while the remaining eight genes are reverse strand. Their lengths range from 64 to 71 bp. The shortest genes are trnC, trnY, trnF, and trnS2, and the longest gene is trnK. There are three tRNA clusters in R. binoculata, which are trnM-trnI-trnQ, trnW-trnC-trnY, and trnA-trnR-trnN-trnS1-trnE-trnF. In general, the R. binoculata mitogenome exhibits a structure congruent with the ancestral Pancrustacea model [47]. Given that the arrangement of genes in gene clusters is different in some species, this is one of the classification characteristics employed to distinguish between species. The gene order of R. binoculata is the same as that of other Sphingidae species and is typical of Lepidoptera [27,38]. Of the two rRNA genes, the large one is rrn16 (1350 bp), which is between trnL1 and trnV, and the small one is rrn12 (778 bp), which is located between trnV and the A+T-rich region. Both of the genes are reverse strand, which is identical to those in other sequenced hawkmoth genomes [27].
In the mitogenome of R. binoculata, there are a total of 8 overlapping regions involving 15 genes, and both the 3′ and 5′ ends of trnT overlap with two other genes. The largest region is 11 bp and is located between the 3′ end of ND4L and the 5′ end of trnT. The smallest overlaps, which are 1 bp, occur between the 3′ end of trnT and the 5′ end of trnP, the 3′ end of trnK and the 5′ end of trnD, and the 3′ end of ND6 and the 5′ end of CYTB. Other overlapping regions are between the 3′ end of trnI and the 5′ end of trnQ, between the 3′ end of trnW and the 5′ end of trnC, between the 3′ end of ATP8 and the 5′ end of ATP6, and between the 3′ end of ND3 and the 5′ end of trnA, with a length of 3, 8, 7, and 2 bp, respectively. The overlap sequence 5′-ATGATAA-3′ between ATP8 and ATP6 is commonly found in all known Lepidopteran mitochondrial genomes [38,43,48,49].
The AT-rich region, which is also known as the control region, is located between rrn12 and trnM and has an average length of 439 bp [50]. The length of the control region in the mitogenome of R. binoculata is 425 bp. This region is considered to be associated with the origin of replication and transcription [24,47]. This region is characterized by a high content of adenine and thymine, with the control region of R. binoculata having an A+T content of 92.48%. Discrepancies in the start and stop codons suggest the potential divergence between R. binoculata and R. mongoliana. Phylogenetic analysis indicates that R. binoculata and R. mongoliana are clustered together within the Macroglossinae subfamily, emphasizing their similarity in evolution. Although the phylogenetic trees analyzed using MrBayes and IQ-TREE have identical structures, indicating a certain level of reliability, some nodes have branch support values where ML is less than 50 and PP is less than 0.5. This suggests that the classification of these branches is not very stable. It is hoped that with the release of more genetic sequencing data, these issues can be resolved in the future.
In summary, this study provides crucial genetic insights into the endemic moth species R. binoculata, offering a foundation for future research on its ecological and molecular aspects. Our comparative genomics and phylogenetic analysis contribute valuable resources for broader studies on the evolutionary dynamics and biodiversity within the Sphingidae family. In the future, the practical application of R. binoculata in the BEVS will undergo further testing and evaluation.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/genes15091171/s1: Supplementary Figure S1: Coverage plot of R. binoculata mitogenome; Supplementary Table S1: Primer sets in this study; Supplementary Table S2: The mitogenome sequence used in this study.
Author Contributions
Conceptualization, Y.-S.N. and T.-Y.W.; methodology, Y.-Y.K., J.-C.C., Y.-H.L., and Y.-F.H.; validation, Y.-Y.K., T.-Y.W., and Y.-S.N.; formal analysis, Y.-Y.K., J.-C.C., Y.-H.L., and Y.-F.H.; data curation, Y.-Y.K. and Y.-S.N.; writing—original draft preparation, Y.-Y.K.; writing—review and editing, Y.-Y.K., T.-Y.W., and Y.-S.N.; visualization, Y.-Y.K. and Y.-S.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Grant 109-2313-B-005-048 -MY3 from the National Science and Technology Council (NSTC) and NSTC 110-2313-B-033-001-.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The sequencing raw data were deposited in the NCBI (https://www.ncbi.nlm.nih.gov, accessed on 28 November 2023) database with the Sequence Read Archive (SRA) accession number PRJNA1045710. The mitogenome data are available in the NCBI database under the GenBank reference number OQ812083.1.
Acknowledgments
We thank Jing-Fu Tsai and Bao-Cheng Lai from the National Museum of Natural Science (NMNS) for kindly helping and providing us with the adult specimen of R. binoculata.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- van Nieukerken, E.J.; Kitching, I.J.; Kristensen, N.P.; Lees, D.C.; Minet, J.; Mitter, C.; Mutanen, M.; Regier, J.C.; Simonsen, T.J.; Wahlberg, N.; et al. Order Lepidoptera Linnaeus; 1758. Zhang, Z.-Q. Animal biodiversity: An outline of higher-level classification and survey of taxonomic richness. Zootaxa 2011, 3148, 212–221. Available online: https://mapress.com/zt/article/view/zootaxa.3148.1.41 (accessed on 3 September 2024). [CrossRef]
- Regier, J.C.; Zwick, A.; Cummings, M.P.; Kawahara, A.Y.; Cho, S.; Weller, S.; Roe, A.; Baixeras, J.; Brown, J.W.; Parr, C.; et al. Toward reconstructing the evolution of advanced moths and butterflies (Lepidoptera: Ditrysia): An initial molecular study. BMC Evol. Biol. 2009, 9, 280. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, H.; Liu, Y.D.; Liu, S.S.; Xia, J.; Zhang, K.; Geng, J. Organization and phylogenetic relationships of the mitochondrial genomes of and other lepidopteran insects. Sci. Rep. 2021, 11, 2957. [Google Scholar] [CrossRef]
- Sun, Y.-X.; Wang, L.; Wei, G.-Q.; Qian, C.; Dai, L.-S.; Sun, Y.; Abbas, M.N.; Zhu, B.-J.; Liu, C.-L. Characterization of the complete mitochondrial genome of Leucoma salicis (Lepidoptera: Lymantriidae) and comparison with other lepidopteran insects. Sci. Rep. 2016, 6, 39153. [Google Scholar] [CrossRef]
- Wei, Z.-X.; Sun, G.; Shiu, J.-Y.; Fang, Y.; Shi, Q.-H. The complete mitochondrial genome sequence of Dodona eugenes (Lepidoptera: Riodinidae). Mitochondrial DNA Part B 2021, 6, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Kabiraj, D.; Sharma, P.; Chetia, H.; Mosahari, P.V.; Neog, K.; Bora, U. The mitochondrial genome of Muga silkworm (Antheraea assamensis) and its comparative analysis with other lepidopteran insects. PLoS ONE 2017, 12, e0188077. [Google Scholar] [CrossRef]
- Chen, D.B.; Zhang, R.S.; Jin, X.D.; Yang, J.; Li, P.; Liu, Y.Q. First complete mitochondrial genome of Rhodinia species (Lepidoptera: Saturniidae): Genome description and phylogenetic implication. Bull. Èntomol. Res. 2022, 112, 243–252. [Google Scholar] [CrossRef]
- Liu, X.M.; Qi, M.J.; Xu, H.Z.; Wu, Z.P.; Hu, L.Z.; Yang, M.S.; Li, H.H. Nine Mitochondrial Genomes of the Pyraloidea and Their Phylogenetic Implications (Lepidoptera). Insects 2021, 12, 1039. [Google Scholar] [CrossRef]
- Xu, C.; Pan, Z.-Q.; Nie, L.; Hao, J.-S. The complete mitochdrial genome of Issoria eugenia (Lepidoptera: Nymphalidae: Heliconiinae). Mitochondrial DNA Part B 2019, 4, 1662–1663. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, C.; Gao, J.; Abbas, M.N.; Kausar, S.; Qian, C.; Wang, L.; Wei, G.; Zhu, B.-J.; Liu, C.-L. Comparative mitochondrial genome analysis of Daphnis nerii and other lepidopteran insects reveals conserved mitochondrial genome organization and phylogenetic relationships. PLoS ONE 2017, 12, e0178773. [Google Scholar] [CrossRef]
- Huang, Y.-X.; Xing, Z.-P.; Zhang, H.; Xu, Z.-B.; Tao, L.-L.; Hu, H.-Y.; Kitching, I.J.; Wang, X. Characterization of the complete mitochondrial genome of eight diurnal hawkmoths (Lepidoptera: Sphingidae): New insights into the origin and evolution of diurnalism in sphingids. Insects 2022, 13, 887. [Google Scholar] [CrossRef] [PubMed]
- Rhagastis binoculata Matsumura, 1909. The Digital Museum of Nature & Culture. Available online: http://catalog.digitalarchives.tw/item/00/11/46/41.html (accessed on 28 September 2023).
- Jiang, Z.-H.; Wang, J.-X.; Xu, Z.-B.; Kitching, I.J.; Huang, C.-L.; Hu, S.-J.; Xiao, Y.-L. Revision of the Genus Rhagastis Rothschild & Jordan, 1903 (Lepidoptera: Sphingidae) from China, Based on Morphological and Phylogenetic Analyses. Insects 2024, 15, 359. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.-T.; Fan, Y.-B.; Warneke, A. Larger Moths (Lepidoptera: Bombycoidea and Sphingoidea) of the Fushan Experimental Forest, Northern Taiwan. Taiwan J. For. Sci. 1999, 14, 469–478. [Google Scholar]
- Jehle, J.A.; Blissard, G.; Bonning, B.; Cory, J.; Herniou, E.; Rohrmann, G.; Theilmann, D.; Thiem, S.; Vlak, J. On the classification and nomenclature of baculoviruses: A proposal for revision. Arch. Virol. 2006, 151, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Huang, C.-W.; Nai, Y.-S.; Chu, P.-Y.; Wang, C.-H.; Ding, S.-T. A newly designed EGFP-2A peptide monocistronic baculoviral vector for concatenating the expression of recombinant proteins in insect cells. Processes 2019, 7, 291. [Google Scholar] [CrossRef]
- Cao, C.; Wu, X.; Zhao, N.; Yao, H.; Lu, X.; Tan, Y. Development of a rapid and efficient BmNPV baculovirus expression system for application in mulberry silkworm, Bombyx mori. Curr. Sci. 2006, 91, 1692–1697. [Google Scholar]
- Song, J.; Wang, X.; Hou, D.; Huang, H.; Liu, X.; Deng, F.; Wang, H.; Arif, B.M.; Hu, Z.; Wang, M. The host specificities of baculovirus per os infectivity factors. PLoS ONE 2016, 11, e0159862. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Li, D.H.; Liu, C.M.; Luo, R.B.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-S.; Wang, X.; Meng, Y.-F.; Huang, Y.-X. Complete mitochondrial characteristics and phylogenetic analysis of Deilephila elpenor (Linnaeus, 1758) (Lepidoptera: Sphingidae). Mitochondrial DNA Part B 2023, 8, 952–955. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Hu, K.; Zhao, Y.; Lin, R.; Li, Y.; Huang, Z.; Zhang, X.; Geng, X.; Ding, J. Mitochondrial genome characteristics of two Sphingidae insects (Psilogramma increta and Macroglossum stellatarum) and implications for their phylogeny. Int. J. Biol. Macromol. 2018, 113, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Kitching, I.; Xu, Z.-B.; Huang, Y.-X. First mitogenome of subfamily Langiinae (Lepidoptera: Sphingidae) with its phylogenetic implications. Gene 2021, 789, 145667. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Qu, W.-Y.; Huang, Y.-X. The complete mitochondrial genome sequence of the hawk moth, Theretra oldenlandiae (Lepidoptera: Sphingidae). Mitochondrial DNA Part B 2020, 5, 978–979. [Google Scholar] [CrossRef]
- Lu, Q.; Yao, H.; Zhang, J.; Xu, H.; Jiang, C. The complete mitogenome sequence of the hawk moth, Theretra latreillii subsp. lucasii (Lepidoptera: Sphingidae) from Zhejiang Province, China. Mitochondrial DNA Part B 2021, 6, 1880–1882. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat. Commun. 2022, 13, 6968. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Audic, S.; Claverie, J.-M.; Blanc, G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol. Biol. 2010, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M. Phylogeny. fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, L.; Liao, C.-Q.; Wang, X.; Wang, M.; Huang, G.-H. Comparative mitochondrial genome analysis and phylogenetic relationship among lepidopteran species. Gene 2022, 830, 146516. [Google Scholar] [CrossRef]
- Liu, D.; Basso, A.; Babbucci, M.; Patarnello, T.; Negrisolo, E. Macrostructural Evolution of the Mitogenome of Butterflies (Lepidoptera, Papilionoidea). Insects 2022, 13, 358. [Google Scholar] [CrossRef]
- Cao, Y.-Q.; Ma, C.; Chen, J.-Y.; Yang, D.-R. The complete mitochondrial genomes of two ghost moths, Thitarodes renzhiensis and Thitarodes yunnanensis: The ancestral gene arrangement in Lepidoptera. BMC Genom. 2012, 13, 276. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Zhang, X.; Xu, S.; Yang, T.; Yanagimoto, T.; Gao, T. Comparative analysis of the complete mitochondrial genomes of three rockfishes (Scorpaeniformes, Sebastiscus) and insights into the phylogenetic relationships of Sebastidae. Biosci. Rep. 2020, 40, BSR20203379. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Y.; Lin, R.; Zhang, Y.; Hu, K.; Li, Y.; Huang, Z.; Peng, S.; Ding, J.; Geng, X. Mitochondrial genome characteristics of Somena scintillans (Lepidoptera: Erebidae) and comparation with other Noctuoidea insects. Genomics 2019, 111, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Dai, J.; Gao, Q.; Yuan, G.; Liu, J.; Sun, Y.; Sun, Y.; Wang, L.; Qian, C.; Zhu, B. Characterization of the complete mitochondrial genome of Orthaga olivacea Warre (Lepidoptera Pyralidae) and comparison with other Lepidopteran insects. PLoS ONE 2020, 15, e0227831. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.-N.; Chai, X.-Y.; Bian, D.-D.; Zhou, C.-L.; Tang, B.-P. The complete mitochondrial genome of Plodia interpunctella (Lepidoptera: Pyralidae) and comparison with other Pyraloidea insects. Genome 2016, 59, 37–49. [Google Scholar] [CrossRef]
- Li, Q.; Wang, X.; Chen, X.; Han, B. Complete mitochondrial genome of the tea looper caterpillar, Ectropis obliqua (Lepidoptera: Geometridae) with a phylogenetic analysis of Geometridae. Int. J. Biol. Macromol. 2018, 114, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Dong, W.-W.; Dong, S.-Y.; Jiang, G.-F.; Huang, G.-H. Characterization of the complete mitochondrial genome of tea tussock moth, Euproctis pseudoconspersa (Lepidoptera: Lymantriidae) and its phylogenetic implications. Gene 2016, 577, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-F.; Su, T.-J.; Luo, A.-R.; Zhu, C.-D.; Wu, C.-S. Characterization of the complete mitochondrion genome of diurnal moth Amata emma (Butler)(Lepidoptera: Erebidae) and its phylogenetic implications. PLoS ONE 2013, 8, e72410. [Google Scholar] [CrossRef]
- Taylor, M.; McKechnie, S.W.; Pierce, N.; Kreitman, M. The lepidopteran mitochondrial control region: Structure and evolution. Mol. Biol. Evol. 1993, 10, 1259–1272. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).