
The Role of RNA Splicing in Liver Function and Disease: A Focus on Metabolic Dysfunction-Associated Steatotic Liver Disease


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Different Types of Splicing
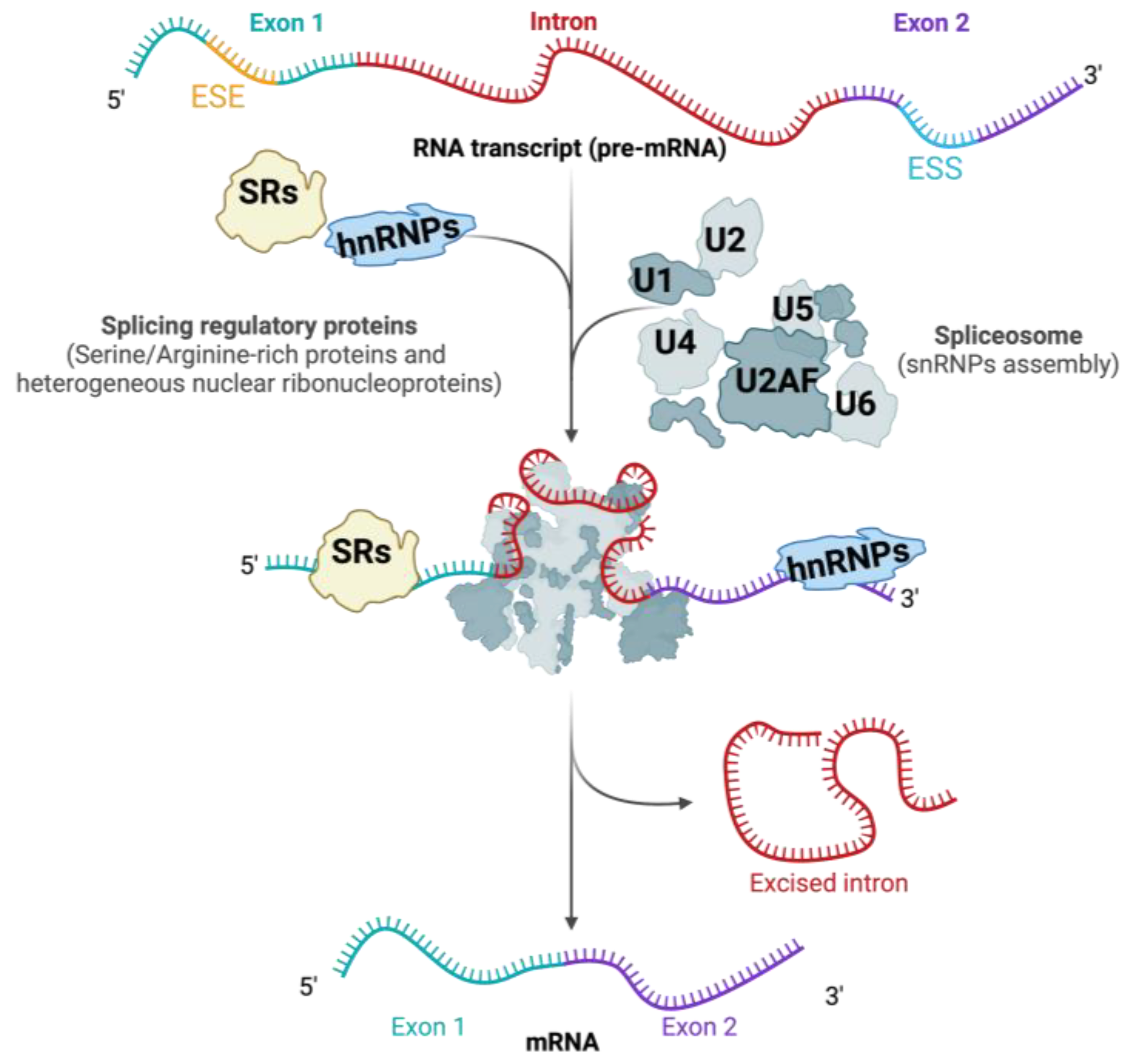
3. RNA Splicing: Mechanisms and Regulation
3.1. Cis-Regulatory Sequences in RNA Splicing
3.2. Trans-Acting Splicing Regulatory Proteins
3.2.1. The Role of Serine-/Arginine-Rich (SR) Proteins
3.2.2. The Role of hnRNPs
3.2.3. The Role of Other RNA-Binding Proteins
4. Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD)
Understanding MASLD: Pathogenesis and Clinical Implications
5. The Role of Alternative Splicing in MASLD
5.1. Cis-Regulatory Sequence Mutations and SNPs in MASLD
5.2. Splicing Regulatory Proteins in MASLD
6. Therapeutic Approaches
7. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Bottger, J. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [PubMed]
- Chow, L.T.; Gelinas, R.E.; Broker, T.R.; Roberts, R.J. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 1977, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef]
- Li, Y.I.; van de Geijn, B.; Raj, A.; Knowles, D.A.; Petti, A.A.; Golan, D.; Gilad, Y.; Pritchard, J.K. RNA splicing is a primary link between genetic variation and disease. Science 2016, 352, 600–604. [Google Scholar] [CrossRef]
- Wong, C.M.; Xu, L.; Yau, M.Y. Alternative mRNA Splicing in the Pathogenesis of Obesity. Int. J. Mol. Sci. 2018, 19, 632. [Google Scholar] [CrossRef] [PubMed]
- Naing, Y.T.; Sun, L. The Role of Splicing Factors in Adipogenesis and Thermogenesis. Mol. Cells 2023, 46, 268–277. [Google Scholar] [CrossRef]
- Webster, N.J.G.; Kumar, D.; Wu, P. Dysregulation of RNA splicing in early non-alcoholic fatty liver disease through hepatocellular carcinoma. Sci. Rep. 2024, 14, 2500. [Google Scholar] [CrossRef]
- Cui, D.; Wang, Z.; Dang, Q.; Wang, J.; Qin, J.; Song, J.; Zhai, X.; Zhou, Y.; Zhao, L.; Lu, G.; et al. Spliceosome component Usp39 contributes to hepatic lipid homeostasis through the regulation of autophagy. Nat. Commun. 2023, 14, 7032. [Google Scholar] [CrossRef]
- Allen, A.M.; Therneau, T.M.; Larson, J.J.; Coward, A.; Somers, V.K.; Kamath, P.S. Nonalcoholic fatty liver disease incidence and impact on metabolic burden and death: A 20 year-community study. Hepatology 2018, 67, 1726–1736. [Google Scholar] [CrossRef]
- Rehati, A.; Abuduaini, B.; Liang, Z.; Chen, D.; He, F. Identification of heat shock protein family A member 5 (HSPA5) targets involved in nonalcoholic fatty liver disease. Genes. Immun. 2023, 24, 124–129. [Google Scholar] [CrossRef]
- Wang, Y.; Song, L.; Ning, M.; Hu, J.; Cai, H.; Song, W.; Gong, D.; Liu, L.; Smith, J.; Li, H.; et al. Identification of alternative splicing events related to fatty liver formation in duck using full-length transcripts. BMC Genomics 2023, 24, 92. [Google Scholar] [CrossRef]
- van den Hoogenhof, M.M.; Pinto, Y.M.; Creemers, E.E. RNA Splicing: Regulation and Dysregulation in the Heart. Circ. Res. 2016, 118, 454–468. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xu, Q.G.; Wang, Z.G.; Yang, Y.; Zhang, L.; Ma, J.Z.; Sun, S.H.; Yang, F.; Zhou, W.P. Circular RNA cSMARCA5 inhibits growth and metastasis in hepatocellular carcinoma. J. Hepatol. 2018, 68, 1214–1227. [Google Scholar] [CrossRef]
- Wang, G.; Tong, J.; Li, Y.; Qiu, X.; Chen, A.; Chang, C.; Yu, G. Overview of CircRNAs Roles and Mechanisms in Liver Fibrosis. Biomolecules 2023, 13, 940. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Behzadnia, N.; Golas, M.M.; Hartmuth, K.; Sander, B.; Kastner, B.; Deckert, J.; Dube, P.; Will, C.L.; Urlaub, H.; Stark, H.; et al. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J. 2007, 26, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef]
- Jeong, S. SR Proteins: Binders, Regulators, and Connectors of RNA. Mol. Cells 2017, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jobbins, A.M.; Campagne, S.; Weinmeister, R.; Lucas, C.M.; Gosliga, A.R.; Clery, A.; Chen, L.; Eperon, L.P.; Hodson, M.J.; Hudson, A.J.; et al. Exon-independent recruitment of SRSF1 is mediated by U1 snRNP stem-loop 3. EMBO J. 2022, 41, e107640. [Google Scholar] [CrossRef] [PubMed]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef]
- Bogaert, E.; Garde, A.; Gautier, T.; Rooney, K.; Duffourd, Y.; LeBlanc, P.; van Reempts, E.; Tran Mau-Them, F.; Wentzensen, I.M.; Au, K.S.; et al. SRSF1 haploinsufficiency is responsible for a syndromic developmental disorder associated with intellectual disability. Am. J. Hum. Genet. 2023, 110, 790–808. [Google Scholar] [CrossRef]
- Lei, S.; Zhang, B.; Huang, L.; Zheng, Z.; Xie, S.; Shen, L.; Breitzig, M.; Czachor, A.; Liu, H.; Luo, H.; et al. SRSF1 promotes the inclusion of exon 3 of SRA1 and the invasion of hepatocellular carcinoma cells by interacting with exon 3 of SRA1pre-mRNA. Cell Death Discov. 2021, 7, 117. [Google Scholar] [CrossRef]
- Cheng, Y.; Luo, C.; Wu, W.; Xie, Z.; Fu, X.; Feng, Y. Liver-Specific Deletion of SRSF2 Caused Acute Liver Failure and Early Death in Mice. Mol. Cell Biol. 2016, 36, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shen, L.; Yuan, W.; Liu, Y.; Guo, R.; Luo, Y.; Zhan, Z.; Xie, Z.; Wu, G.; Wu, W.; et al. Loss of SRSF2 triggers hepatic progenitor cell activation and tumor development in mice. Commun. Biol. 2020, 3, 210. [Google Scholar] [CrossRef]
- Hosono, N. Genetic abnormalities and pathophysiology of MDS. Int. J. Clin. Oncol. 2019, 24, 885–892. [Google Scholar] [CrossRef]
- Luo, C.; Cheng, Y.; Liu, Y.; Chen, L.; Liu, L.; Wei, N.; Xie, Z.; Wu, W.; Feng, Y. SRSF2 Regulates Alternative Splicing to Drive Hepatocellular Carcinoma Development. Cancer Res. 2017, 77, 1168–1178. [Google Scholar] [CrossRef]
- Sen, S.; Jumaa, H.; Webster, N.J. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat. Commun. 2013, 4, 1336. [Google Scholar] [CrossRef]
- Du, K.; Peng, Y.; Greenbaum, L.E.; Haber, B.A.; Taub, R. HRS/SRp40-mediated inclusion of the fibronectin EIIIB exon, a possible cause of increased EIIIB expression in proliferating liver. Mol. Cell Biol. 1997, 17, 4096–4104. [Google Scholar] [CrossRef]
- Jensen, M.A.; Wilkinson, J.E.; Krainer, A.R. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat. Struct. Mol. Biol. 2014, 21, 189–197. [Google Scholar] [CrossRef]
- Juan-Mateu, J.; Alvelos, M.I.; Turatsinze, J.V.; Villate, O.; Lizarraga-Mollinedo, E.; Grieco, F.A.; Marroqui, L.; Bugliani, M.; Marchetti, P.; Eizirik, D.L. SRp55 Regulates a Splicing Network That Controls Human Pancreatic beta-Cell Function and Survival. Diabetes 2018, 67, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, J.; Lu, Y.; Bian, H.; Yang, L.; Wu, H.; Zhang, X.; Zhang, B.; Xiong, M.; Chang, Y.; et al. DRAK2 aggravates nonalcoholic fatty liver disease progression through SRSF6-associated RNA alternative splicing. Cell Metab. 2021, 33, 2004–2020.e2009. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Delannoy, A.; Salvetti, A.; Chabot, B. SRSF10: An atypical splicing regulator with critical roles in stress response, organ development, and viral replication. RNA 2021, 27, 1302–1317. [Google Scholar] [CrossRef]
- Feng, Y.; Valley, M.T.; Lazar, J.; Yang, A.L.; Bronson, R.T.; Firestein, S.; Coetzee, W.A.; Manley, J.L. SRp38 regulates alternative splicing and is required for Ca(2+) handling in the embryonic heart. Dev. Cell 2009, 16, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Cheng, Y.; Wang, Z.; Liu, Y.; Luo, C.; Liu, L.; Chen, L.; Xie, Z.; Lu, Y.; Feng, Y. SRSF10 Plays a Role in Myoblast Differentiation and Glucose Production via Regulation of Alternative Splicing. Cell Rep. 2015, 13, 1647–1657. [Google Scholar] [CrossRef]
- Elliott, D.J.; Best, A.; Dalgliesh, C.; Ehrmann, I.; Grellscheid, S. How does Tra2beta protein regulate tissue-specific RNA splicing? Biochem. Soc. Trans. 2012, 40, 784–788. [Google Scholar] [CrossRef]
- Pihlajamaki, J.; Lerin, C.; Itkonen, P.; Boes, T.; Floss, T.; Schroeder, J.; Dearie, F.; Crunkhorn, S.; Burak, F.; Jimenez-Chillaron, J.C.; et al. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab. 2011, 14, 208–218. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, J.; Manley, J.L. Turning on a fuel switch of cancer: hnRNP proteins regulate alternative splicing of pyruvate kinase mRNA. Cancer Res. 2010, 70, 8977–8980. [Google Scholar] [CrossRef]
- Talukdar, I.; Sen, S.; Urbano, R.; Thompson, J.; Yates, J.R., 3rd; Webster, N.J. hnRNP A1 and hnRNP F modulate the alternative splicing of exon 11 of the insulin receptor gene. PLoS ONE 2011, 6, e27869. [Google Scholar] [CrossRef] [PubMed]
- Zarnack, K.; Konig, J.; Tajnik, M.; Martincorena, I.; Eustermann, S.; Stevant, I.; Reyes, A.; Anders, S.; Luscombe, N.M.; Ule, J. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 2013, 152, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Luo, X.; Xie, M.; Zhang, T.; Chen, X.; Zhang, B.; Sun, M.; Wang, Y.; Feng, Y.; Ji, X.; et al. HNRNPC downregulation inhibits IL-6/STAT3-mediated HCC metastasis by decreasing HIF1A expression. Cancer Sci. 2022, 113, 3347–3361. [Google Scholar] [CrossRef]
- Singh, A.B.; Li, H.; Kan, C.F.; Dong, B.; Nicolls, M.R.; Liu, J. The critical role of mRNA destabilizing protein heterogeneous nuclear ribonucleoprotein d in 3′ untranslated region-mediated decay of low-density lipoprotein receptor mRNA in liver tissue. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Alcedo, K.P.; Kim, H.J.; Snider, N.T. Alternative Splicing in Hepatocellular Carcinoma. Cell Mol. Gastroenterol. Hepatol. 2020, 10, 699–712. [Google Scholar] [CrossRef]
- Chang, J.J.; Lin, T.; Jhang, X.Y.; Chan, S.P. hnRNP Q/SYNCRIP interacts with LIN28B and modulates the LIN28B/let-7 axis in human hepatoma cells. PLoS ONE 2024, 19, e0304947. [Google Scholar] [CrossRef]
- Yuan, J.H.; Liu, X.N.; Wang, T.T.; Pan, W.; Tao, Q.F.; Zhou, W.P.; Wang, F.; Sun, S.H. The MBNL3 splicing factor promotes hepatocellular carcinoma by increasing PXN expression through the alternative splicing of lncRNA-PXN-AS1. Nat. Cell Biol. 2017, 19, 820–832. [Google Scholar] [CrossRef]
- Sen, S.; Talukdar, I.; Liu, Y.; Tam, J.; Reddy, S.; Webster, N.J. Muscleblind-like 1 (Mbnl1) promotes insulin receptor exon 11 inclusion via binding to a downstream evolutionarily conserved intronic enhancer. J. Biol. Chem. 2010, 285, 25426–25437. [Google Scholar] [CrossRef]
- Kim, S.E.; Park, C.K.; Park, J.W.; Lee, J.W.; Choe, J.Y.; Cho, Y.A. Clinicopathologic Significance of Quaking Expression in Hepatocellular Carcinoma. In Vivo 2024, 38, 2064–2073. [Google Scholar] [CrossRef]
- Paterson, H.A.B.; Yu, S.; Artigas, N.; Prado, M.A.; Haberman, N.; Wang, Y.F.; Jobbins, A.M.; Pahita, E.; Mokochinski, J.; Hall, Z.; et al. Liver RBFOX2 regulates cholesterol homeostasis via Scarb1 alternative splicing in mice. Nat. Metab. 2022, 4, 1812–1829. [Google Scholar] [CrossRef]
- Bhate, A.; Parker, D.J.; Bebee, T.W.; Ahn, J.; Arif, W.; Rashan, E.H.; Chorghade, S.; Chau, A.; Lee, J.H.; Anakk, S.; et al. ESRP2 controls an adult splicing programme in hepatocytes to support postnatal liver maturation. Nat. Commun. 2015, 6, 8768. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Pathophysiological Molecular Mechanisms of Obesity: A Link between MAFLD and NASH with Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 11629. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Cuevas, J.; Lucano-Landeros, S.; Lopez-Cifuentes, D.; Santos, A.; Armendariz-Borunda, J. Epidemiologic, Genetic, Pathogenic, Metabolic, Epigenetic Aspects Involved in NASH-HCC: Current Therapeutic Strategies. Cancers 2022, 15, 23. [Google Scholar] [CrossRef]
- Clark, J.M. The epidemiology of nonalcoholic fatty liver disease in adults. J. Clin. Gastroenterol. 2006, 40 (Suppl. S1), S5–S10. [Google Scholar]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Ratziu, V.; Bellentani, S.; Cortez-Pinto, H.; Day, C.; Marchesini, G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J Hepatol 2010, 53, 372–384. [Google Scholar] [CrossRef]
- Duell, P.B.; Welty, F.K.; Miller, M.; Chait, A.; Hammond, G.; Ahmad, Z.; Cohen, D.E.; Horton, J.D.; Pressman, G.S.; Toth, P.P.; et al. Nonalcoholic Fatty Liver Disease and Cardiovascular Risk: A Scientific Statement From the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e168–e185. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Hui, S.T.; Kurt, Z.; Tuominen, I.; Norheim, F.; Davis, R.; Pan, C.; Dirks, D.L.; Magyar, C.E.; French, S.W.; Chella Krishnan, K.; et al. The Genetic Architecture of Diet-Induced Hepatic Fibrosis in Mice. Hepatology 2018, 68, 2182–2196. [Google Scholar] [CrossRef]
- Hui, S.T.; Parks, B.W.; Org, E.; Norheim, F.; Che, N.; Pan, C.; Castellani, L.W.; Charugundla, S.; Dirks, D.L.; Psychogios, N.; et al. The genetic architecture of NAFLD among inbred strains of mice. Elife 2015, 4, e05607. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Baker, S.S.; Moylan, C.A.; Abdelmalek, M.F.; Guy, C.D.; Zamboni, F.; Wu, D.; Lin, W.; Liu, W.; Baker, R.D.; et al. Systematic transcriptome analysis reveals elevated expression of alcohol-metabolizing genes in NAFLD livers. J. Pathol. 2016, 238, 531–542. [Google Scholar] [CrossRef]
- Correia, J.C.; Massart, J.; de Boer, J.F.; Porsmyr-Palmertz, M.; Martinez-Redondo, V.; Agudelo, L.Z.; Sinha, I.; Meierhofer, D.; Ribeiro, V.; Bjornholm, M.; et al. Bioenergetic cues shift FXR splicing towards FXRalpha2 to modulate hepatic lipolysis and fatty acid metabolism. Mol. Metab. 2015, 4, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pastor, A.R.; Gomez-Hernandez, A.; Diaz-Castroverde, S.; Gonzalez-Aseguinolaza, G.; Gonzalez-Rodriguez, A.; Garcia, G.; Fernandez, S.; Escribano, O.; Benito, M. Liver-specific insulin receptor isoform A expression enhances hepatic glucose uptake and ameliorates liver steatosis in a mouse model of diet-induced obesity. Dis. Model. Mech. 2019, 12, dmm036186. [Google Scholar] [CrossRef]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Reviejo, M.; Soto, M.; Lozano, E.; Asensio, M.; Ortiz-Rivero, S.; Berasain, C.; Avila, M.A.; Herraez, E. Impact of Alternative Splicing Variants on Liver Cancer Biology. Cancers 2021, 14, 18. [Google Scholar] [CrossRef]
- Xu, K.; Wu, T.; Xia, P.; Chen, X.; Yuan, Y. Alternative splicing: A bridge connecting NAFLD and HCC. Trends Mol. Med. 2023, 29, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Marsh, S.; Hu, J.; Feng, W.; Wu, C. The Pathogenesis of Nonalcoholic Fatty Liver Disease: Interplay between Diet, Gut Microbiota, and Genetic Background. Gastroenterol. Res. Pract. 2016, 2016, 2862173. [Google Scholar] [CrossRef]
- Grander, C.; Grabherr, F.; Tilg, H. Non-alcoholic fatty liver disease: Pathophysiological concepts and treatment options. Cardiovasc. Res. 2023, 119, 1787–1798. [Google Scholar] [CrossRef]
- Sharma, D.; Mandal, P. NAFLD: Genetics and its clinical implications. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 102003. [Google Scholar] [CrossRef]
- Wang, P.; Wu, C.X.; Li, Y.; Shen, N. HSD17B13 rs72613567 protects against liver diseases and histological progression of nonalcoholic fatty liver disease: A systematic review and meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8997–9007. [Google Scholar] [PubMed]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Pirola, C.J.; Sookoian, S.; Wilson, L.A.; Liang, T.; Chalasani, N. The Protection Conferred by HSD17B13 rs72613567 Polymorphism on Risk of Steatohepatitis and Fibrosis May Be Limited to Selected Subgroups of Patients With NAFLD. Clin. Transl. Gastroenterol. 2021, 12, e00400. [Google Scholar] [CrossRef]
- Ratziu, V.; Lalazar, A.; Wong, L.; Dang, Q.; Collins, C.; Shaulian, E.; Jensen, S.; Friedman, S.L. Zf9, a Kruppel-like transcription factor up-regulated in vivo during early hepatic fibrosis. Proc. Natl. Acad. Sci. USA 1998, 95, 9500–9505. [Google Scholar] [CrossRef]
- Starkel, P.; Sempoux, C.; Leclercq, I.; Herin, M.; Deby, C.; Desager, J.P.; Horsmans, Y. Oxidative stress, KLF6 and transforming growth factor-beta up-regulation differentiate non-alcoholic steatohepatitis progressing to fibrosis from uncomplicated steatosis in rats. J. Hepatol. 2003, 39, 538–546. [Google Scholar] [CrossRef]
- Miele, L.; Beale, G.; Patman, G.; Nobili, V.; Leathart, J.; Grieco, A.; Abate, M.; Friedman, S.L.; Narla, G.; Bugianesi, E.; et al. The Kruppel-like factor 6 genotype is associated with fibrosis in nonalcoholic fatty liver disease. Gastroenterology 2008, 135, 282–291.e1. [Google Scholar] [CrossRef]
- Narla, G.; DiFeo, A.; Yao, S.; Banno, A.; Hod, E.; Reeves, H.L.; Qiao, R.F.; Camacho-Vanegas, O.; Levine, A.; Kirschenbaum, A.; et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005, 65, 5761–5768. [Google Scholar] [CrossRef]
- Kim, M.J.; Yu, C.Y.; Theusch, E.; Naidoo, D.; Stevens, K.; Kuang, Y.L.; Schuetz, E.; Chaudhry, A.S.; Medina, M.W. SUGP1 is a novel regulator of cholesterol metabolism. Hum. Mol. Genet. 2016, 25, 3106–3116. [Google Scholar] [CrossRef] [PubMed]
- Del Rio-Moreno, M.; Alors-Perez, E.; Gonzalez-Rubio, S.; Ferrin, G.; Reyes, O.; Rodriguez-Peralvarez, M.; Sanchez-Frias, M.E.; Sanchez-Sanchez, R.; Ventura, S.; Lopez-Miranda, J.; et al. Dysregulation of the Splicing Machinery Is Associated to the Development of Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2019, 104, 3389–3402. [Google Scholar] [CrossRef]
- Arif, W.; Mathur, B.; Saikali, M.F.; Chembazhi, U.V.; Toohill, K.; Song, Y.J.; Hao, Q.; Karimi, S.; Blue, S.M.; Yee, B.A.; et al. Splicing factor SRSF1 deficiency in the liver triggers NASH-like pathology and cell death. Nat. Commun. 2023, 14, 551. [Google Scholar] [CrossRef]
- Sen, S.; Langiewicz, M.; Jumaa, H.; Webster, N.J. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology 2015, 61, 171–183. [Google Scholar] [CrossRef]
- Kumar, D.; Das, M.; Sauceda, C.; Ellies, L.G.; Kuo, K.; Parwal, P.; Kaur, M.; Jih, L.; Bandyopadhyay, G.K.; Burton, D.; et al. Degradation of splicing factor SRSF3 contributes to progressive liver disease. J. Clin. Investig. 2019, 129, 4477–4491. [Google Scholar] [CrossRef]
- Dalamaga, M.; Liu, J. DRAK2-SRSF6-regulated RNA alternative splicing is a promising therapeutic target in NAFLD/NASH. Metabol. Open 2022, 13, 100157. [Google Scholar] [CrossRef]
- Jobbins, A.M.; Haberman, N.; Artigas, N.; Amourda, C.; Paterson, H.A.B.; Yu, S.; Blackford, S.J.I.; Montoya, A.; Dore, M.; Wang, Y.F.; et al. Dysregulated RNA polyadenylation contributes to metabolic impairment in non-alcoholic fatty liver disease. Nucleic Acids Res. 2022, 50, 3379–3393. [Google Scholar] [CrossRef]
- Xiong, J.; Liu, T.; Mi, L.; Kuang, H.; Xiong, X.; Chen, Z.; Li, S.; Lin, J.D. hnRNPU/TrkB Defines a Chromatin Accessibility Checkpoint for Liver Injury and Nonalcoholic Steatohepatitis Pathogenesis. Hepatology 2020, 71, 1228–1246. [Google Scholar] [CrossRef]
- Kim, K.; Choi, S.H. A New Modality in Dyslipidemia Treatment: Antisense Oligonucleotide Therapy. J. Lipid Atheroscler. 2022, 11, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.M.; Balfour, J.A. Fomivirsen. Drugs 1999, 57, 375–380; discussion 381. [Google Scholar] [CrossRef] [PubMed]
- Chambergo-Michilot, D.; Alur, A.; Kulkarni, S.; Agarwala, A. Mipomersen in Familial Hypercholesterolemia: An Update on Health-Related Quality of Life and Patient-Reported Outcomes. Vasc. Health Risk Manag. 2022, 18, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Corey, D.R. The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy. Nucleic Acid. Ther. 2020, 30, 67–70. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Wurster, C.D.; Ludolph, A.C. Nusinersen for spinal muscular atrophy. Ther. Adv. Neurol. Disord. 2018, 11, 1756285618754459. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Inotersen: First Global Approval. Drugs 2018, 78, 1371–1376. [Google Scholar] [CrossRef]
- Blair, H.A. Tofersen: First Approval. Drugs 2023, 83, 1039–1043. [Google Scholar] [CrossRef]
- Paik, J.; Duggan, S. Volanesorsen: First Global Approval. Drugs 2019, 79, 1349–1354. [Google Scholar] [CrossRef]
- Prohaska, T.A.; Alexander, V.J.; Karwatowska-Prokopczuk, E.; Tami, J.; Xia, S.; Witztum, J.L.; Tsimikas, S. APOC3 inhibition with volanesorsen reduces hepatic steatosis in patients with severe hypertriglyceridemia. J. Clin. Lipidol. 2023, 17, 406–411. [Google Scholar] [CrossRef]
- Lopez-Canovas, J.L.; Del Rio-Moreno, M.; Garcia-Fernandez, H.; Jimenez-Vacas, J.M.; Moreno-Montilla, M.T.; Sanchez-Frias, M.E.; Amado, V.; Fernando, L.; Fondevila, M.F.; Ciria, R.; et al. Splicing factor SF3B1 is overexpressed and implicated in the aggressiveness and survival of hepatocellular carcinoma. Cancer Lett. 2021, 496, 72–83. [Google Scholar] [CrossRef]
- da Silva, M.R.; Moreira, G.A.; Goncalves da Silva, R.A.; de Almeida Alves Barbosa, E.; Pais Siqueira, R.; Teixera, R.R.; Almeida, M.R.; Silva Junior, A.; Fietto, J.L.; Bressan, G.C. Splicing Regulators and Their Roles in Cancer Biology and Therapy. Biomed. Res. Int. 2015, 2015, 150514. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaminska, D. The Role of RNA Splicing in Liver Function and Disease: A Focus on Metabolic Dysfunction-Associated Steatotic Liver Disease. Genes 2024, 15, 1181. https://doi.org/10.3390/genes15091181
Kaminska D. The Role of RNA Splicing in Liver Function and Disease: A Focus on Metabolic Dysfunction-Associated Steatotic Liver Disease. Genes. 2024; 15(9):1181. https://doi.org/10.3390/genes15091181
Chicago/Turabian StyleKaminska, Dorota. 2024. "The Role of RNA Splicing in Liver Function and Disease: A Focus on Metabolic Dysfunction-Associated Steatotic Liver Disease" Genes 15, no. 9: 1181. https://doi.org/10.3390/genes15091181
APA StyleKaminska, D. (2024). The Role of RNA Splicing in Liver Function and Disease: A Focus on Metabolic Dysfunction-Associated Steatotic Liver Disease. Genes, 15(9), 1181. https://doi.org/10.3390/genes15091181