
A Novel KIDINS220 Pathogenic Variant Associated with the Syndromic Spastic Paraplegia SINO: An Expansion of the Brain Malformation Spectrum and a Literature Review

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Brain Imaging
2.3. Genetic Analysis
2.4 Literature Review
3. Results
3.1. Clinical Report
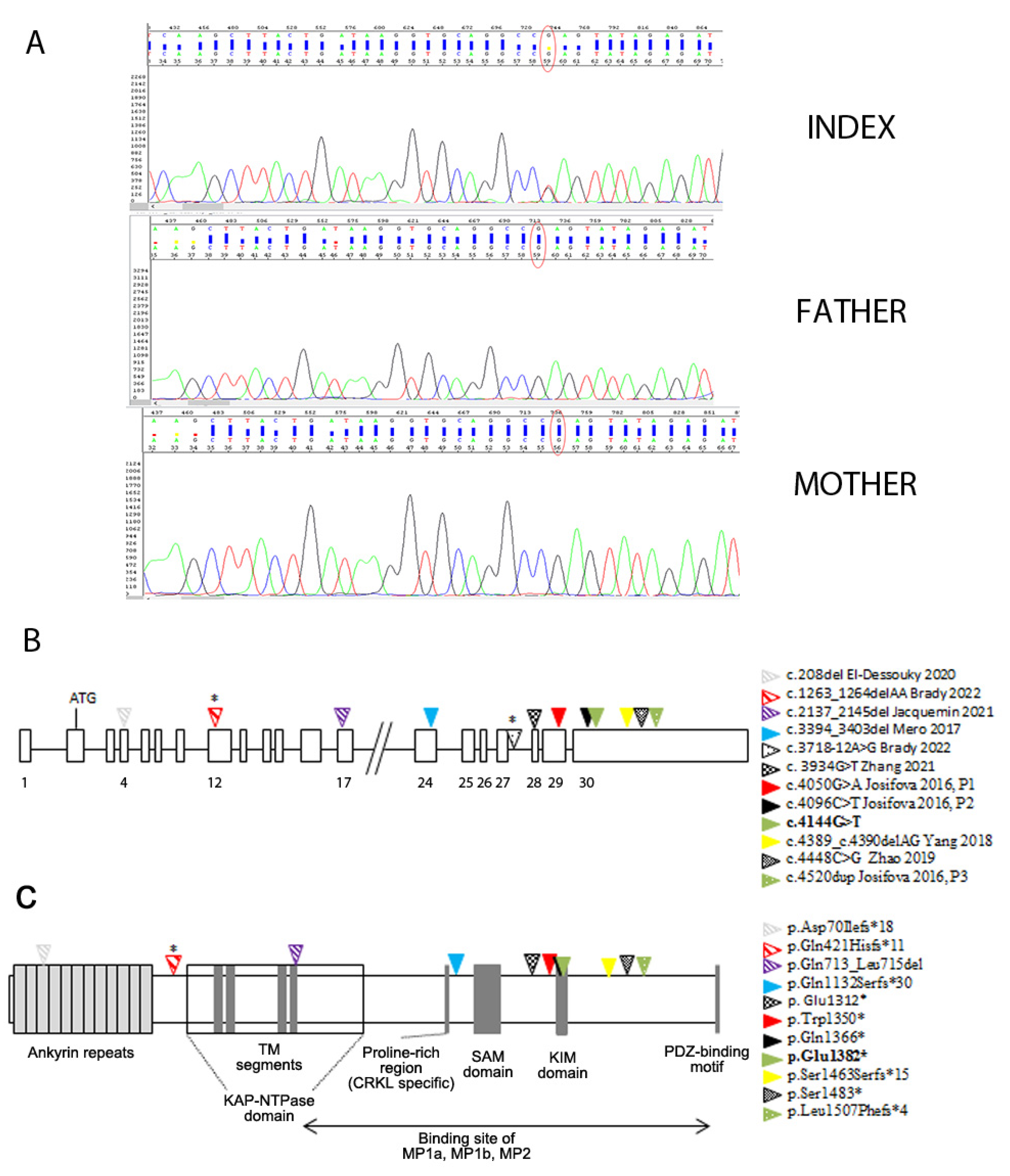
3.2. Genetic Findings
3.3. Spectrum of Brain Malformation in Prenatal Cases and Postnatal Patients Affected by SINO
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arévalo, J.C.; Yano, H.; Teng, K.K.; Chao, M.V. A unique pathway for sustained neurotrophin signaling through an ankyrin-rich membrane-spanning protein. EMBO J. 2004, 23, 2358–2368. [Google Scholar] [CrossRef]
- Bracale, A.; Cesca, F.; Neubrand, V.E.; Newsome, T.P.; Way, M.; Schiavo, G. Kidins220/ARMS is transported by a kinesin-1-based mechanism likely to be involved in neuronal differentiation. Mol. Biol. Cell 2007, 18, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Higuero, A.M.; Sánchez-Ruiloba, L.; Doglio, L.E.; Portillo, F.; Abad-Rodríguez, J.; Dotti, C.G.; Iglesias, T. Kidins220/ARMS modulates the activity of microtubule-regulating proteins and controls neuronal polarity and development. J. Biol. Chem. 2010, 285, 1343–1357. [Google Scholar] [CrossRef] [PubMed]
- Scholz-Starke, J.; Cesca, F.; Schiavo, G.; Benfenati, F.; Baldelli, P. Kidins220/ARMS is a novel modulator of short-term synaptic plasticity in hippocampal GABAergic neurons. PLoS ONE 2012, 7, e35785. [Google Scholar] [CrossRef]
- Neubrand, V.E.; Cesca, F.; Benfenati, F.; Schiavo, G. Kidins220/ARMS as a functional mediator of multiple receptor signalling pathways. J. Cell Sci. 2012, 125 Pt 8, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Josifova, D.J.; Monroe, G.R.; Tessadori, F.; de Graaff, E.; van der Zwaag, B.; Mehta, S.G.; Harakalova, M.; Duran, K.J.; Savelberg, S.M.; DDD Study. Heterozygous KIDINS220/ARMS nonsense variants cause spastic paraplegia, intellectual disability, nystagmus, and obesity. Hum. Mol. Genet. 2016, 25, 2158–2167. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, W.; Peng, J.; Yin, F. Heterozygous KIDINS220 mutation leads to spastic paraplegia and obesity in an Asian girl. Eur. J. Neurol. 2018, 25, e53–e54. [Google Scholar] [CrossRef]
- Zhang, K.; Sun, W.; Liu, Y.; Lv, Y.; Hou, D.; Lin, Y.; Xu, W.; Zhao, J.; Gai, Z.; Zhao, S.; et al. SINO Syndrome Causative KIDINS220/ARMS Gene Regulates Adipocyte Differentiation. Front. Cell Dev. Biol. 2021, 9, 619475. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, Y.J.; Wang, M.W.; Lin, X.H.; Dong, E.L.; Chen, W.J.; Wang, N.; Lin, X. Genetic and Clinical Profile of Chinese Patients with Autosomal Dominant Spastic Paraplegia. Mol. Diagn. Ther. 2019, 23, 781–789. [Google Scholar] [CrossRef]
- Mero, I.L.; Mørk, H.H.; Sheng, Y.; Blomhoff, A.; Opheim, G.L.; Erichsen, A.; Vigeland, M.D.; Selmer, K.K. Homozygous KIDINS220 loss-of-function variants in fetuses with cerebral ventriculomegaly and limb contractures. Hum. Mol. Genet. 2017, 26, 3792–3796. [Google Scholar] [CrossRef]
- Jacquemin, V.; Antoine, M.; Duerinckx, S.; Massart, A.; Desir, J.; Perazzolo, C.; Cassart, M.; Thomas, D.; Segers, V.; Lecomte, S.; et al. TrkA mediates effect of novel KIDINS220 mutation in human brain ventriculomegaly. Hum. Mol. Genet. 2021, 29, 3757–3764. [Google Scholar] [CrossRef] [PubMed]
- El-Dessouky, S.H.; Issa, M.Y.; Aboulghar, M.M.; Gaafar, H.M.; Elarab, A.E.; Ateya, M.I.; Omar, H.H.; Beetz, C.; Zaki, M.S. Prenatal delineation of a distinct lethal fetal syndrome caused by a homozygous truncating KIDINS220 variant. Am. J. Med. Genet. A 2020, 182, 2867–2876. [Google Scholar] [CrossRef]
- Brady, L.I.; DeFrance, B.; Tarnopolsky, M. Pre- and Postnatal Characterization of Autosomal Recessive KIDINS220-Associated Ventriculomegaly. Mol. Syndromol. 2022, 13, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Pezzani, L.; Marchetti, D.; Cereda, A.; Caffi, L.G.; Manara, O.; Mamoli, D.; Pezzoli, L.; Lincesso, A.R.; Perego, L.; Pellicioli, I.; et al. Atypical presentation of pediatric BRAF RASopathy with acute encephalopathy. Am. J. Med. Genet. A 2018, 176, 2867–2871. [Google Scholar] [CrossRef] [PubMed]
- CinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 18 August 2024).
- HGMD® Professional. Available online: https://apps.ingenuity.com/ingsso/login (accessed on 18 August 2024).
- PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/ (accessed on 5 August 2024).
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef]
- Turner, T.N.; Wilfert, A.B.; Bakken, T.E.; Bernier, R.A.; Pepper, M.R.; Zhang, Z.; Torene, R.I.; Retterer, K.; Eichler, E.E. Sex-Based Analysis of De Novo Variants in Neurodevelopmental Disorders. Am. J. Hum. Genet. 2019, 105, 1274–1285. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Baris, H.N.; Wu, C.; Rudolph, G.; Van Maldergem, L.; He, W.; Chan, W.M.; Andrews, C.; Demer, J.L.; Robertson, R.L.; et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 2010, 140, 74–87. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Cederquist, G.Y.; Gupta Jr, M.L.; Engle, E.C. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr. Opin. Genet. Dev. 2011, 21, 286–294. [Google Scholar] [CrossRef]
- Poirier, K.; Saillour, Y.; Bahi-Buisson, N.; Jaglin, X.H.; Fallet-Bianco, C.; Nabbout, R.; Castelnau-Ptakhine, L.; Roubertie, A.; Attie-Bitach, T.; Desguerre, I.; et al. Mutations in the neuronal ß-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum. Mol. Genet. 2010, 19, 4462–4473. [Google Scholar] [CrossRef]
- Fallet-Bianco, C.; Laquerrière, A.; Poirier, K.; Razavi, F.; Guimiot, F.; Dias, P.; Loeuillet, L.; Lascelles, K.; Beldjord, C.; Carion, N.; et al. Mutations in tubulin genes are frequent causes of various foetal malformations of cortical development including microlissencephaly. Acta Neuropathol. Commun. 2014, 2, 69. [Google Scholar] [CrossRef]
- Gonçalves, F.G.; Freddi, T.A.L.; Taranath, A.; Lakshmanan, R.; Goetti, R.; Feltrin, F.S.; Mankad, K.; Teixeira, S.R.; Hanagandi, P.B.; Arrigoni, F. Tubulinopathies. Top. Magn. Reson. Imaging 2018, 27, 395–408. [Google Scholar] [CrossRef]
- Arrigoni, F.; Romaniello, R.; Peruzzo, D.; Poretti, A.; Bassi, M.T.; Pierpaoli, C.; Valente, E.M.; Nuovo, S.; Boltshauser, E.; Huisman, T.A.G.M.; et al. The spectrum of brainstem malformations associated to mutations of the tubulin genes family: MRI and DTI analysis. Eur. Radiol. 2019, 29, 770–782. [Google Scholar] [CrossRef] [PubMed]
- GeneReviews®–NCBI Bookshel. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1116/ (accessed on 5 July 2024).
- Orphanet. Available online: https://www.orpha.net/ (accessed on 10 June 2024).
- OMIM. Available online: https://www.ncbi.nlm.nih.gov/omim/ (accessed on 1 August 2024).
- Schmieg, N.; Thomas, C.; Yabe, A.; Lynch, D.S.; Iglesias, T.; Chakravarty, P.; Schiavo, G. Novel Kidins220/ARMS Splice Isoforms: Potential Specific Regulators of Neuronal and Cardiovascular Development. PLoS ONE 2015, 10, e0129944. [Google Scholar] [CrossRef] [PubMed]
- Albini, M.; Almacellas-Barbanoj, A.; Krawczun-Rygmaczewska, A.; Ciano, L.; Benfenati, F.; Michetti, C.; Cesca, F. Alterations in KIDINS220/ARMS Expression Impact Sensory Processing and Social Behavior in Adult Mice. Int. J. Mol. Sci. 2024, 25, 2334. [Google Scholar] [CrossRef]
- Cesca, F.; Schiavo, G.; Benfenati, F. Kidins220/ARMS transgenic lines could be instrumental in the understanding of the molecular mechanisms leading to spastic paraplegia and obesity. Eur. J. Neurol. 2018, 25, e107. [Google Scholar] [CrossRef] [PubMed]
- Almacellas-Barbanoj, A.; Albini, M.; Satapathy, A.; Jaudon, F.; Michetti, C.; Krawczun-Rygmaczewska, A.; Huang, H.; Manago, F.; Papaleo, F.; Benfenati, F.; et al. Kidins220/ARMS modulates brain morphology and anxiety-like traits in adult mice. Cell Death Discov. 2022, 8, 58. [Google Scholar] [CrossRef]
- Adle-Biassette, H.; Saugier-Veber, P.; Fallet-Bianco, C.; Delezoide, A.L.; Razavi, F.; Drouot, N.; Bazin, A.; Beaufrère, A.M.; Bessières, B.; Blesson, S.; et al. Neuropathological review of 138 cases genetically tested for X-linked hydrocephalus: Evidence for closely related clinical entities of unknown molecular bases. Acta Neuropathol. 2013, 126, 427–442. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| KIDINS220 (NM_020738.4) Variants | Inheritance | Prenatal US (WG) | Age at Brain MRI | Brain MRI | Age at Last Follow-Up Gender | Neurological/ Postmortem Examination | ID | Growth Parameters | Craniofacial Dysmorphisms | References |
|---|---|---|---|---|---|---|---|---|---|---|
| c. 208del; p.Asp70Ilefs*18 (exon 4) | AR (homozygous) | Dilated ventricles (3/3), CC agenesis (3/3), cerebellar hypoplasia (2/3), cerebellar vermis agenesis (2/3) ↑NT (1/3), CHD (3/3), hydrops fetalis (2/3),polyhydramnios (2/3), ascites (1/3) Normal for gestational age (1/3); NA (2/3) (12, 22 and 24) | / | NP | / 1M, 2F | Limb contractures (3/3) | / | / | Brachyplagiocephaly (3/3), bossed forehead (3/3), deep set eyes (3/3), micrognathia (3/3) | [12] |
| c.1263_1264delAA p.Gln421Hisfs*11 (exon 12) pat c.3718-12A>G (inron 27) mat | AR (compound heterozygous) | Bilateral ventriculomegaly (19 +3) Further ventricle enlargement, bilateral talipes equinovarus (20 +3) Polyhydramnios (31) Large for gestational age (35 + 3) | 21 and 27 WG 8th day | 3rd ventricle dilatation, thin corpus callosum, possible absence of the cavum septum pellucidum Thin corpus callosum, thin brainstem, absence of the cavum septum pellucidum, hypoplasia of the basal ganglia, thalami, and inferior cerebellar vermis | 2.5 y F | Coarse nystagmus SP | Severe DD At last follow-up, minimal head control, unable to sit, reach, or grab for items, vocalizations only | COF>99th p, weight at 90th p at birth COF>98th p, length<3rd p, weight at 60th p at 18 m | Frontal bossing, mild micrognathia | [13] |
| c.2137-2145del p.Gln713_Leu715del (exon 17) | AR (homozygous) Parents’ first-degree cousins | Severe ventriculomegaly (3/3) (14 and second-semester) Clenched hands, bent wrists, club feet (3/3) Normal for gestational age (3/3) | Post Mortem (P in 1/3) | Triventricular hydrocephalus, cortical atrophy without gyri (lissencephaly), confirmed at autopsy | / 2M, 1F | Limb contractures (3/3) | / | / | - | [11] |
| c.3394_3403del; p.Gln1132Serfs*30 (exon 24) | AR (homozygous) | Hydrocephalus/dilated ventricles (2/3), CC agenesis (1/3) NA growth parameters (13, 1 Fe; 18, 2 Fe) | / | NP | / NA | Limb contractures (3/3) | / | / | Micrognathia (1/3) | [10] |
| c.3934G>T; p.Glu1312* (exon 28) | AD | NA | / | NP | 39 y F | Nystagmus, SP | Moderate ID (IQ 39 at WAIS) | Severe obesity (BMI 35.6 kg/m2) | - | [8], mother |
| NA | / | NP | 17 y M | Nystagmus, SP | Moderate ID (IQ 42 at WAIS) | Obesity (BMI 29.4 kg/m2) | Brachycephaly | [8], elder son | ||
| Normal findings | 19 m | Normal findings | 5 y M | SP | Moderate ID | Early-onset overgrowth (macrocephaly, height and weight >99th p), obesity | Brachycephaly | [8], younger son | ||
| c.4050G>A; p.Trp1350* (exon 29) | de novo | Dilated lateral ventricles (23) | 12 m | Dilated 3rd and lateral ventricles, ↓ WM bulk, mild delay in myelination, mild generalized atrophy | 14 y M | Nystagmus, axial hypotonia, SP | Moderate ID; he speaks in sentences since 4 y | Early-onset overgrowth (OFC, height and weight >90th p) | Brachyplagiocephaly, bossed forehead, deep-set eyes | [6], patient 1 |
| c.4096C>T; p.Gln1366* (exon 30) | de novo | Dilated lateral ventricles (20) | 24 m | Dilated 3rd and lateral ventricles, ↓ WM bulk, mild generalized atrophy | 15 y M | Nystagmus, squint, SP | Moderate ID; he speaks in sentences since 4 y | Early-onset overgrowth (OFC, height and weight >90th p) | Brachyplagiocephaly, prominent forehead, deep-set eyes, crowded teeth | [6], patient 2 |
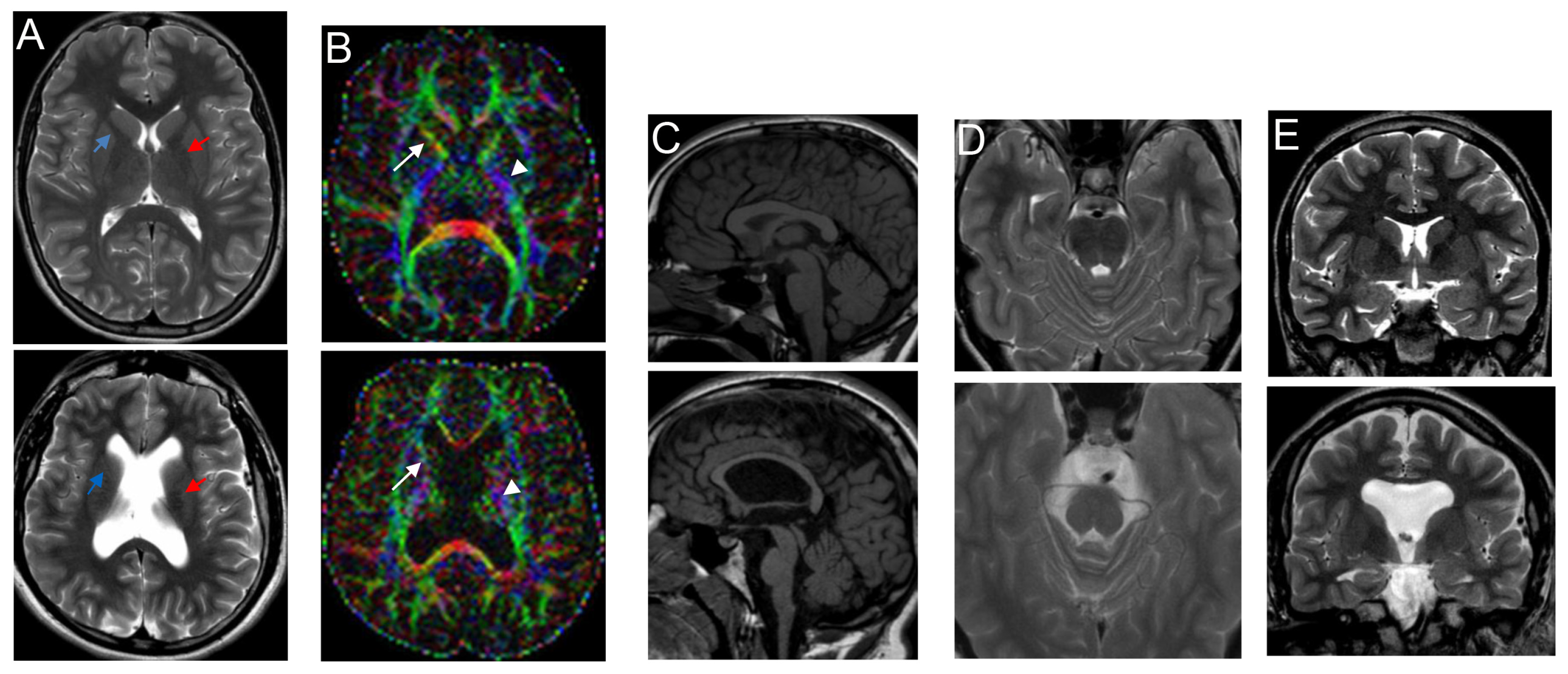
| c.4144G>T; p.Glu1382* (exon 30) | de novo | Unilateral ventriculomegaly (3rd trimester) | 22 m | Lateral ventricle enlargement, pellucidum septum agenesis, CC hypoplasia, verrticalized hippocampi, thin and dysmorphic brainstem | 18 y M | Monolateral squint, SP | Severe ID; he speaks in sentences | Early-onset macrocephaly, obesity | High forehead | Present study |
| 17 y | Also, dysmorphic and hypoplastic ALIC, partial fusion of lenticular and caudate nuclei, corticospinal tract thinning, optic chiasm hypoplasia, ↓ frontal and temporal lobe volume with cortical gyration simplification, absence of superior cerebellar peduncles decussation | |||||||||
| c.4389_c.4390delAG; p.Ser1463Serfs*15 (exon 30) | de novo | Normal findings | 3 y | Periventricular WM high signals at FLAIR, ↓ splenium and posterior corpus callosi, chiasma opticum, and optic nerve hypoplasia | 3 y F | SP | Mild ID | Obesity | Short philtrum, ectropion of nostril, plump cheeks | [7] |
| c.4448C>G; p.Ser1483* (exon 30) | AD | NA | / | NP (5/5) | NA 3M, 2F | SP (5/5) | - (5/5) | Normal (5/5) | - (5/5) | [9] |
| c.4520dup; p.Leu1507Phefs*4 (exon 30) | de novo | Dilated lateral ventricles | 30m | High-riding 3rd ventricle, dilated lateral ventricles, partial CC agenesis | 7 y M | Nystagmus, squint, SP | Moderate ID; few single words by 24 m | Early-onset obesity | Brachyplagiocephaly, prominent forehead | [6], patient 3; The variant has been also reported in a patient with NDD [18,19] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonati, M.T.; Baldoli, C.; Taurino, J.; Marchetti, D.; Larizza, L.; Finelli, P.; Iascone, M. A Novel KIDINS220 Pathogenic Variant Associated with the Syndromic Spastic Paraplegia SINO: An Expansion of the Brain Malformation Spectrum and a Literature Review. Genes 2024, 15, 1190. https://doi.org/10.3390/genes15091190
Bonati MT, Baldoli C, Taurino J, Marchetti D, Larizza L, Finelli P, Iascone M. A Novel KIDINS220 Pathogenic Variant Associated with the Syndromic Spastic Paraplegia SINO: An Expansion of the Brain Malformation Spectrum and a Literature Review. Genes. 2024; 15(9):1190. https://doi.org/10.3390/genes15091190
Chicago/Turabian StyleBonati, Maria Teresa, Cristina Baldoli, Jacopo Taurino, Daniela Marchetti, Lidia Larizza, Palma Finelli, and Maria Iascone. 2024. "A Novel KIDINS220 Pathogenic Variant Associated with the Syndromic Spastic Paraplegia SINO: An Expansion of the Brain Malformation Spectrum and a Literature Review" Genes 15, no. 9: 1190. https://doi.org/10.3390/genes15091190