

Expanding the Mutation Spectrum for Inherited Retinal Diseases
 ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Results
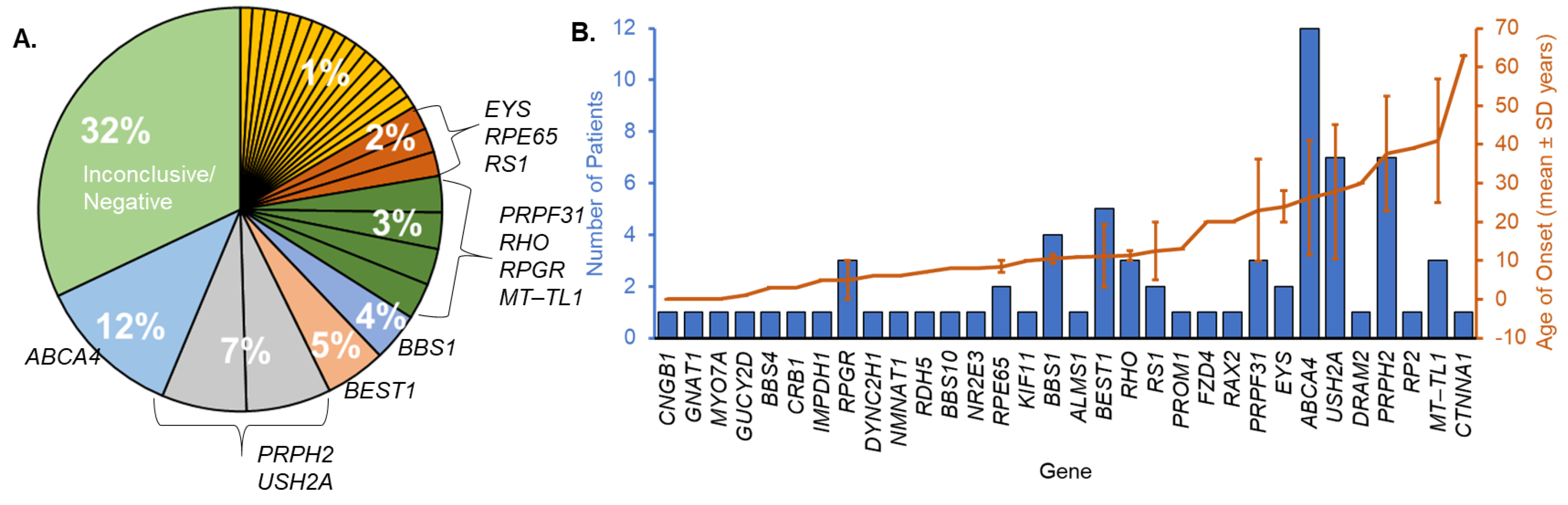
3.1. Cohort Characteristics and Molecular Diagnosis
3.2. Novel Genetics and Clinical Phenotypes
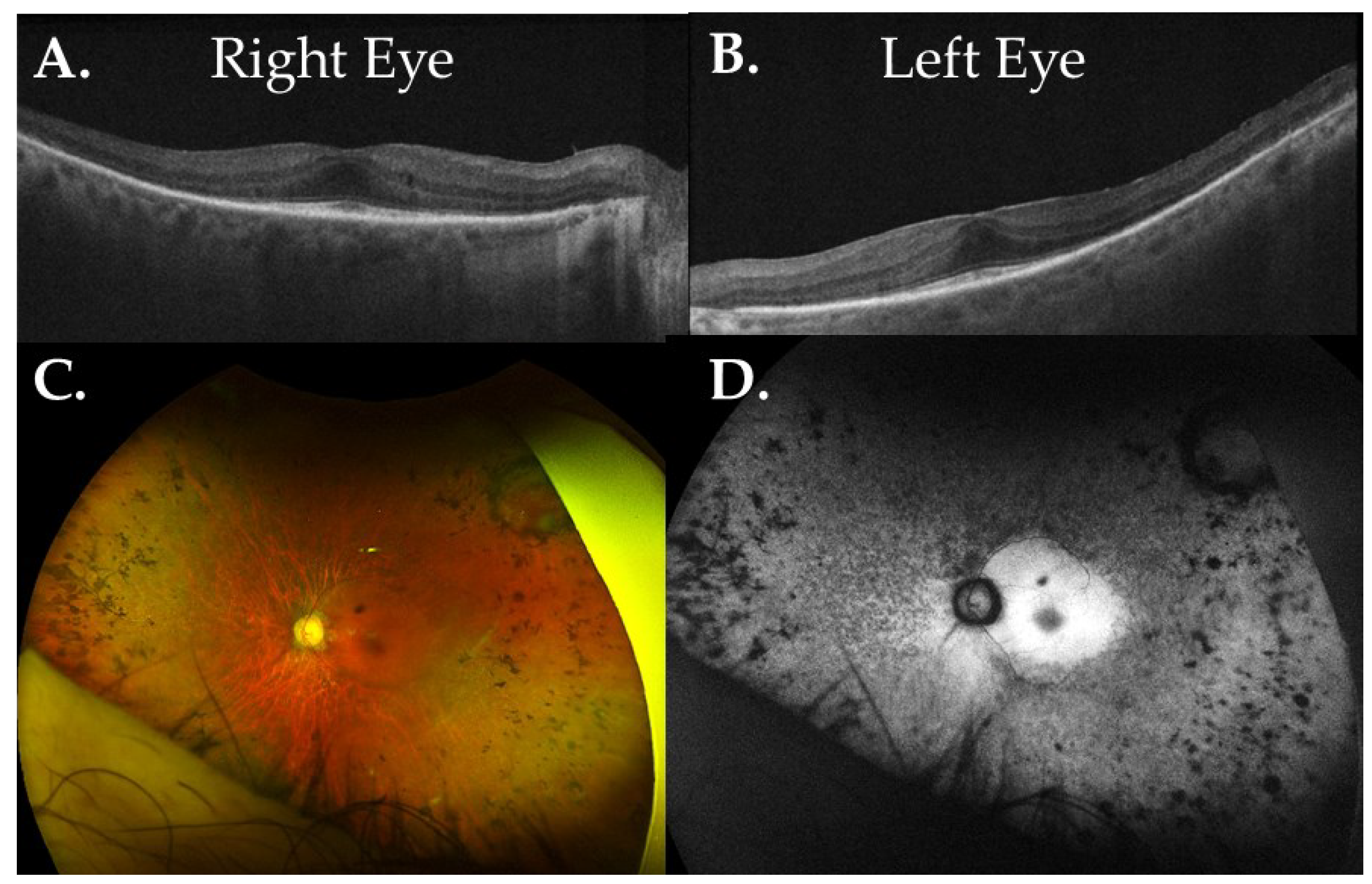
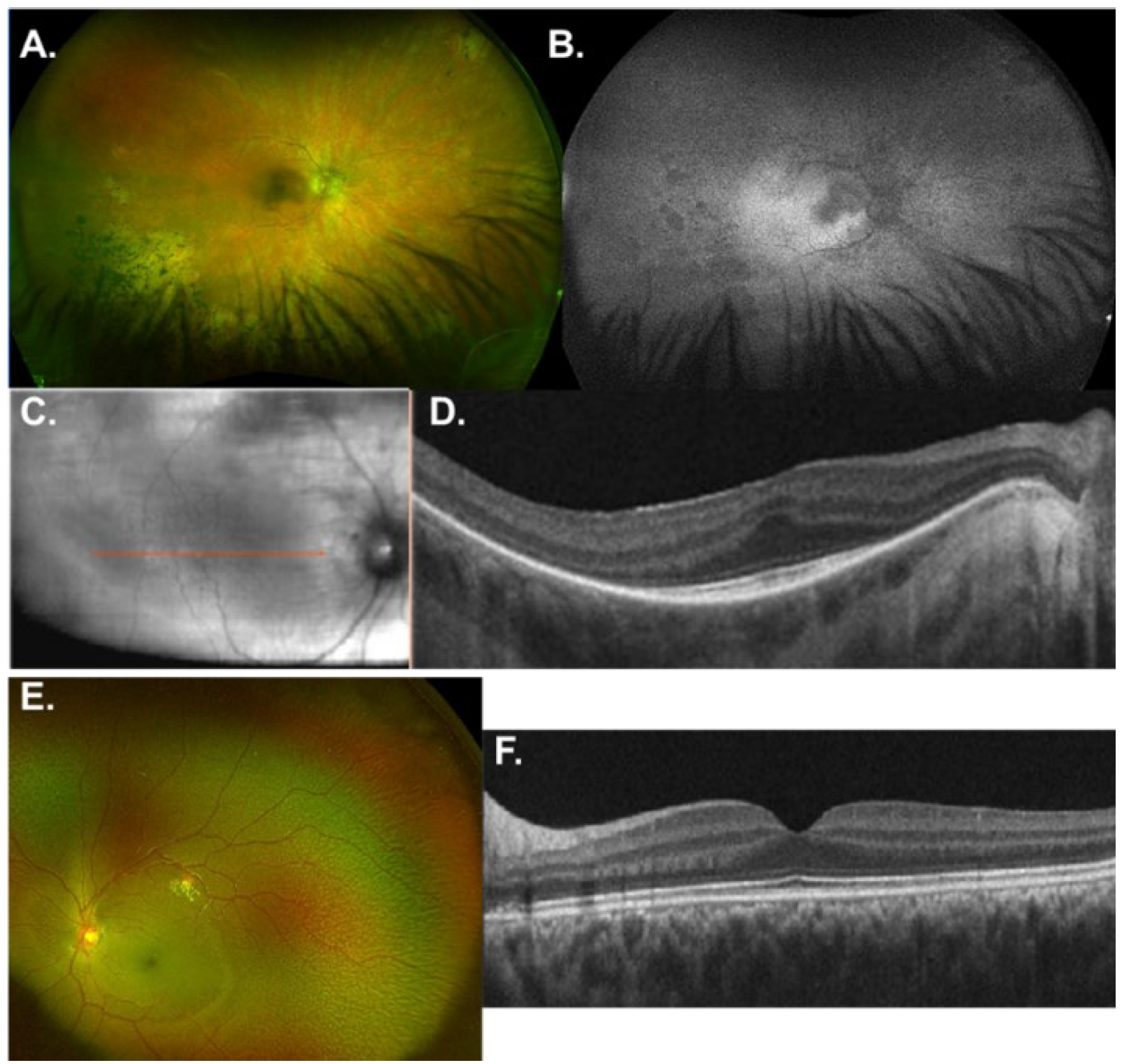
3.2.1. ALMS1
3.2.2. GNAT1
3.2.3. RAX2
3.2.4. RDH5
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bundey, S.; Crews, S.J. A study of retinitis pigmentosa in the City of Birmingham. II Clinical and genetic heterogeneity. J. Med. Genet. 1984, 21, 421–428. [Google Scholar] [CrossRef]
- Hu, D.N. Genetic aspects of retinitis pigmentosa in China. Am. J. Med. Genet. 1982, 12, 51–56. [Google Scholar] [CrossRef]
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Birch, D.G.; Mintz-Hittner, H.; Ruiz, R.S.; Lewis, R.A.; et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001, 17, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013, 84, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Askou, A.L.; Jakobsen, T.S.; Corydon, T.J. Retinal gene therapy: An eye-opener of the 21st century. Gene Ther. 2021, 28, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D.N.; Mimouni, M.; El-Yaniv, R.; Blumenthal, E.Z. Shortening the Early Treatment Diabetic Retinopathy Study visual acuity test utilizing a novel computer software: Reproducibility in control and patient eyes. Acta Ophthalmol. 2021, 99, e1281–e1288. [Google Scholar] [CrossRef]
- Marchant, D.; Yu, K.; Bigot, K.; Roche, O.; Germain, A.; Bonneau, D.; Drouin-Garraud, V.; Schorderet, D.F.; Munier, F.; Schmidt, D.; et al. New VMD2 gene mutations identified in patients affected by Best vitelliform macular dystrophy. J. Med. Genet. 2007, 44, e70. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Yang, G.; Wang, J.; Chen, Y. Screening for BEST1 gene mutations in Chinese patients with bestrophinopathy. Mol. Vis. 2014, 20, 1594–1604. [Google Scholar] [PubMed]
- Bakall, B.; Marknell, T.; Ingvast, S.; Koisti, M.J.; Sandgren, O.; Li, W.; Bergen, A.A.; Andreasson, S.; Rosenberg, T.; Petrukhin, K.; et al. The mutation spectrum of the bestrophin protein--functional implications. Hum. Genet. 1999, 104, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Li, Y.; Kittredge, A.; Hopiavuori, A.; Ward, N.; Yao, P.; Fukuda, Y.; Zhang, Y.; Tsang, S.H.; Yang, T. Investigation and Restoration of BEST1 Activity in Patient-derived RPEs with Dominant Mutations. Sci. Rep. 2019, 9, 19026. [Google Scholar] [CrossRef]
- Frecer, V.; Iarossi, G.; Salvetti, A.P.; Maltese, P.E.; Delledonne, G.; Oldani, M.; Staurenghi, G.; Falsini, B.; Minnella, A.M.; Ziccardi, L.; et al. Pathogenicity of new BEST1 variants identified in Italian patients with best vitelliform macular dystrophy assessed by computational structural biology. J. Transl. Med. 2019, 17, 330. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.H.; Francis, P.J.; Duncan, J.L.; Weleber, R.G.; Saperstein, D.A.; Farrell, D.F.; Stone, E.M. Phenotypic variability due to a novel Glu292Lys variation in exon 8 of the BEST1 gene causing best macular dystrophy. Arch. Ophthalmol. 2009, 127, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Holtan, J.P.; Selmer, K.K.; Heimdal, K.R.; Bragadóttir, R. Inherited retinal disease in Norway—A characterization of current clinical and genetic knowledge. Acta Ophthalmol. 2020, 98, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Y.; Xuan, Y.; Liu, W.; Wang, M. Novel BEST1 Mutations and Special Clinical Features of Best Vitelliform Macular Dystrophy. Ophthalmic Res. 2016, 56, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Stephenson, K.A.J.; Dockery, A.; Turner, J.; O’Byrne, J.J.; Fitzsimon, S.; Farrar, G.J.; Flitcroft, D.I.; Keegan, D.J. Electrophysiology-Guided Genetic Characterisation Maximises Molecular Diagnosis in an Irish Paediatric Inherited Retinal Degeneration Population. Genes 2022, 13, 615. [Google Scholar] [CrossRef]
- Sharon, D.; Ben-Yosef, T.; Goldenberg-Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 2020, 41, 140–149. [Google Scholar] [CrossRef]
- Weisschuh, N.; Obermaier, C.D.; Battke, F.; Bernd, A.; Kuehlewein, L.; Nasser, F.; Zobor, D.; Zrenner, E.; Weber, E.; Wissinger, B.; et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 2020, 41, 1514–1527. [Google Scholar] [CrossRef]
- Preising, M.N.; Pasquay, C.; Friedburg, C.; Bowl, W.; Jäger, M.; Andrassi-Darida, M.; Lorenz, B. [Autosomal recessive bestrophinopathy (ARB): A clinical and molecular description of two patients at childhood]. Klin. Monbl Augenheilkd. 2012, 229, 1009–1017. [Google Scholar] [CrossRef]
- Krämer, F.; Mohr, N.; Kellner, U.; Rudolph, G.; Weber, B.H. Ten novel mutations in VMD2 associated with Best macular dystrophy (BMD). Hum. Mutat. 2003, 22, 418. [Google Scholar] [CrossRef]
- Mykytyn, K.; Nishimura, D.Y.; Searby, C.C.; Shastri, M.; Yen, H.J.; Beck, J.S.; Braun, T.; Streb, L.M.; Cornier, A.S.; Cox, G.F.; et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat. Genet. 2002, 31, 435–438. [Google Scholar] [CrossRef]
- Mykytyn, K.; Nishimura, D.Y.; Searby, C.C.; Beck, G.; Bugge, K.; Haines, H.L.; Cornier, A.S.; Cox, G.F.; Fulton, A.B.; Carmi, R.; et al. Evaluation of complex inheritance involving the most common Bardet-Biedl syndrome locus (BBS1). Am. J. Hum. Genet. 2003, 72, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Beales, P.L.; Badano, J.L.; Ross, A.J.; Ansley, S.J.; Hoskins, B.E.; Kirsten, B.; Mein, C.A.; Froguel, P.; Scambler, P.J.; Lewis, R.A.; et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am. J. Hum. Genet. 2003, 72, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Badano, J.L.; Kim, J.C.; Hoskins, B.E.; Lewis, R.A.; Ansley, S.J.; Cutler, D.J.; Castellan, C.; Beales, P.L.; Leroux, M.R.; Katsanis, N. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 2003, 12, 1651–1659. [Google Scholar] [CrossRef]
- Cox, K.F.; Kerr, N.C.; Kedrov, M.; Nishimura, D.; Jennings, B.J.; Stone, E.M.; Sheffield, V.C.; Iannaccone, A. Phenotypic expression of Bardet-Biedl syndrome in patients homozygous for the common M390R mutation in the BBS1 gene. Vis. Res. 2012, 75, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Gil, N.; Méndez-Vidal, C.; Romero-Pérez, L.; González-del Pozo, M.; Rodríguez-de la Rúa, E.; Dopazo, J.; Borrego, S.; Antiñolo, G. Improving the management of Inherited Retinal Dystrophies by targeted sequencing of a population-specific gene panel. Sci. Rep. 2016, 6, 23910. [Google Scholar] [CrossRef]
- Jespersgaard, C.; Fang, M.; Bertelsen, M.; Dang, X.; Jensen, H.; Chen, Y.; Bech, N.; Dai, L.; Rosenberg, T.; Zhang, J.; et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci. Rep. 2019, 9, 1219. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.; Mullaney, B.G.; Bhaskar, S.S.; Dickerson, J.E.; Hall, G.; O’Grady, A.; Webster, A.; Ramsden, S.C.; Black, G.C. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J. Med. Genet. 2012, 49, 322–326. [Google Scholar] [CrossRef]
- Estrada-Cuzcano, A.; Koenekoop, R.K.; Senechal, A.; De Baere, E.B.; de Ravel, T.; Banfi, S.; Kohl, S.; Ayuso, C.; Sharon, D.; Hoyng, C.B.; et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch. Ophthalmol. 2012, 130, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, Y.; Li, J.; Zhang, W.; Wang, J.; Lewis, R.A.; Wong, L.J. Retinal Diseases Caused by Mutations in Genes Not Specifically Associated with the Clinical Diagnosis. PLoS ONE 2016, 11, e0165405. [Google Scholar] [CrossRef]
- Davis, R.E.; Swiderski, R.E.; Rahmouni, K.; Nishimura, D.Y.; Mullins, R.F.; Agassandian, K.; Philp, A.R.; Searby, C.C.; Andrews, M.P.; Thompson, S.; et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 19422–19427. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Mion, B.; Aherne, A.; Engel, P.C. Molecular recruitment as a basis for negative dominant inheritance? propagation of misfolding in oligomers of IMPDH1, the mutated enzyme in the RP10 form of retinitis pigmentosa. Biochim. Biophys. Acta 2011, 1812, 1472–1476. [Google Scholar] [CrossRef]
- Patel, N.; Aldahmesh, M.A.; Alkuraya, H.; Anazi, S.; Alsharif, H.; Khan, A.O.; Sunker, A.; Al-Mohsen, S.; Abboud, E.B.; Nowilaty, S.R.; et al. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 2016, 18, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Daniels, A.B.; Sandberg, M.A.; Chen, J.; Weigel-DiFranco, C.; Fielding Hejtmancic, J.; Berson, E.L. Genotype-phenotype correlations in Bardet-Biedl syndrome. Arch. Ophthalmol. 2012, 130, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Nasser, F.; Kohl, S.; Kurtenbach, A.; Kempf, M.; Biskup, S.; Zuleger, T.; Haack, T.B.; Weisschuh, N.; Stingl, K.; Zrenner, E. Ophthalmic and Genetic Features of Bardet Biedl Syndrome in a German Cohort. Genes 2022, 13, 1218. [Google Scholar] [CrossRef]
- Kurata, K.; Hosono, K.; Hikoya, A.; Kato, A.; Saitsu, H.; Minoshima, S.; Ogata, T.; Hotta, Y. Clinical characteristics of a Japanese patient with Bardet-Biedl syndrome caused by BBS10 mutations. Jpn. J. Ophthalmol. 2018, 62, 458–466. [Google Scholar] [CrossRef]
- Gerth, C.; Zawadzki, R.J.; Werner, J.S.; Héon, E. Retinal morphology in patients with BBS1 and BBS10 related Bardet-Biedl Syndrome evaluated by Fourier-domain optical coherence tomography. Vis. Res. 2008, 48, 392–399. [Google Scholar] [CrossRef]
- Littink, K.W.; van den Born, L.I.; Koenekoop, R.K.; Collin, R.W.; Zonneveld, M.N.; Blokland, E.A.; Khan, H.; Theelen, T.; Hoyng, C.B.; Cremers, F.P.; et al. Mutations in the EYS gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology 2010, 117, 2026–2033.e7. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Pierrache, L.H.M.; Messchaert, M.; Thiadens, A.; Haer-Wigman, L.; de Jong-Hesse, Y.; van Zelst-Stams, W.A.G.; Collin, R.W.J.; Klaver, C.C.W.; van den Born, L.I. Extending the Spectrum of EYS-Associated Retinal Disease to Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2049–2063. [Google Scholar] [CrossRef]
- Garcia-Delgado, A.B.; Valdes-Sanchez, L.; Morillo-Sanchez, M.J.; Ponte-Zuñiga, B.; Diaz-Corrales, F.J.; de la Cerda, B. Dissecting the role of EYS in retinal degeneration: Clinical and molecular aspects and its implications for future therapy. Orphanet J. Rare Dis. 2021, 16, 222. [Google Scholar] [CrossRef]
- Neubauer, J.; Hahn, L.; Birtel, J.; Boon, C.J.F.; Charbel Issa, P.; Fischer, M.D. GUCY2D-Related Retinal Dystrophy with Autosomal Dominant Inheritance-A Multicenter Case Series and Review of Reported Data. Genes 2022, 13, 313. [Google Scholar] [CrossRef]
- Aguirre, G.K.; Butt, O.H.; Datta, R.; Roman, A.J.; Sumaroka, A.; Schwartz, S.B.; Cideciyan, A.V.; Jacobson, S.G. Postretinal Structure and Function in Severe Congenital Photoreceptor Blindness Caused by Mutations in the GUCY2D Gene. Investig. Ophthalmol. Vis. Sci. 2017, 58, 959–973. [Google Scholar] [CrossRef]
- Liu, X.; Fujinami, K.; Kuniyoshi, K.; Kondo, M.; Ueno, S.; Hayashi, T.; Mochizuki, K.; Kameya, S.; Yang, L.; Fujinami-Yokokawa, Y.; et al. Clinical and Genetic Characteristics of 15 Affected Patients From 12 Japanese Families with GUCY2D-Associated Retinal Disorder. Transl. Vis. Sci. Technol. 2020, 9, 2. [Google Scholar] [CrossRef]
- Kitiratschky, V.B.; Wilke, R.; Renner, A.B.; Kellner, U.; Vadalà, M.; Birch, D.G.; Wissinger, B.; Zrenner, E.; Kohl, S. Mutation analysis identifies GUCY2D as the major gene responsible for autosomal dominant progressive cone degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5015–5023. [Google Scholar] [CrossRef]
- Scopelliti, A.J.; Jamieson, R.V.; Barnes, E.H.; Nash, B.; Rajagopalan, S.; Cornish, E.L.; Grigg, J.R. A natural history study of autosomal dominant GUCY2D-associated cone-rod dystrophy. Doc. Ophthalmol. 2023, 147, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.M.; McClard, C.K.; Mwoyosvi, M.L.; Alkadhem, N.; Radojevic, B.; Klein, M.; Birch, D.; Ellis, A.; Icks, S.W.; Guddanti, T.; et al. Delineating the Clinical Phenotype of Patients With the c.629C>G, p.Pro210Arg Mutation in Peripherin-2. Investig. Ophthalmol. Vis. Sci. 2022, 63, 19. [Google Scholar] [CrossRef]
- Wawrocka, A.; Skorczyk-Werner, A.; Wicher, K.; Niedziela, Z.; Ploski, R.; Rydzanicz, M.; Sykulski, M.; Kociecki, J.; Weisschuh, N.; Kohl, S.; et al. Novel variants identified with next-generation sequencing in Polish patients with cone-rod dystrophy. Mol. Vis. 2018, 24, 326–339. [Google Scholar]
- Renner, A.B.; Fiebig, B.S.; Weber, B.H.; Wissinger, B.; Andreasson, S.; Gal, A.; Cropp, E.; Kohl, S.; Kellner, U. Phenotypic variability and long-term follow-up of patients with known and novel PRPH2/RDS gene mutations. Am. J. Ophthalmol. 2009, 147, 518–530.e1. [Google Scholar] [CrossRef]
- Reeves, M.J.; Goetz, K.E.; Guan, B.; Ullah, E.; Blain, D.; Zein, W.M.; Tumminia, S.J.; Hufnagel, R.B. Genotype-phenotype associations in a large PRPH2-related retinopathy cohort. Hum. Mutat. 2020, 41, 1528–1539. [Google Scholar] [CrossRef]
- Oishi, A.; Fujinami, K.; Mawatari, G.; Naoi, N.; Ikeda, Y.; Ueno, S.; Kuniyoshi, K.; Hayashi, T.; Kondo, H.; Mizota, A.; et al. Genetic and Phenotypic Landscape of PRPH2-Associated Retinal Dystrophy in Japan. Genes 2021, 12, 1817. [Google Scholar] [CrossRef]
- Heath Jeffery, R.C.; Thompson, J.A.; Lamey, T.M.; McLaren, T.L.; De Roach, J.N.; McAllister, I.L.; Constable, I.J.; Chen, F.K. Longitudinal Analysis of Functional and Structural Outcome Measures in PRPH2-Associated Retinal Dystrophy. Ophthalmol. Retin. 2023, 7, 81–91. [Google Scholar] [CrossRef]
- Heath Jeffery, R.C.; Lo, J.; Thompson, J.A.; Lamey, T.M.; McLaren, T.L.; De Roach, J.N.; Ayton, L.N.; Vincent, A.L.; Sharma, A.; Chen, F.K. Analysis of the Outer Retinal Bands in ABCA4 and PRPH2-Associated Retinopathy using OCT. Ophthalmol. Retin. 2024, 8, 174–183. [Google Scholar] [CrossRef]
- Macke, J.P.; Davenport, C.M.; Jacobson, S.G.; Hennessey, J.C.; Gonzalez-Fernandez, F.; Conway, B.P.; Heckenlively, J.; Palmer, R.; Maumenee, I.H.; Sieving, P.; et al. Identification of novel rhodopsin mutations responsible for retinitis pigmentosa: Implications for the structure and function of rhodopsin. Am. J. Hum. Genet. 1993, 53, 80–89. [Google Scholar] [PubMed]
- Fernandez-San Jose, P.; Blanco-Kelly, F.; Corton, M.; Trujillo-Tiebas, M.J.; Gimenez, A.; Avila-Fernandez, A.; Garcia-Sandoval, B.; Lopez-Molina, M.I.; Hernan, I.; Carballo, M.; et al. Prevalence of Rhodopsin mutations in autosomal dominant Retinitis Pigmentosa in Spain: Clinical and analytical review in 200 families. Acta Ophthalmol. 2015, 93, e38–e44. [Google Scholar] [CrossRef] [PubMed]
- Sung, C.H.; Davenport, C.M.; Hennessey, J.C.; Maumenee, I.H.; Jacobson, S.G.; Heckenlively, J.R.; Nowakowski, R.; Fishman, G.; Gouras, P.; Nathans, J. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1991, 88, 6481–6485. [Google Scholar] [CrossRef]
- Roman-Sanchez, R.; Wensel, T.G.; Wilson, J.H. Nonsense mutations in the rhodopsin gene that give rise to mild phenotypes trigger mRNA degradation in human cells by nonsense-mediated decay. Exp. Eye Res. 2016, 145, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Lynn, J.; Raney, A.; Britton, N.; Ramoin, J.; Yang, R.W.; Radojevic, B.; McClard, C.K.; Kingsley, R.; Coussa, R.G.; Bennett, L.D. Genetic Diagnosis for 64 Patients with Inherited Retinal Disease. Genes 2022, 14, 74. [Google Scholar] [CrossRef]
- Miano, M.G.; Testa, F.; Filippini, F.; Trujillo, M.; Conte, I.; Lanzara, C.; Millán, J.M.; De Bernardo, C.; Grammatico, B.; Mangino, M.; et al. Identification of novel RP2 mutations in a subset of X-linked retinitis pigmentosa families and prediction of new domains. Hum. Mutat. 2001, 18, 109–119. [Google Scholar] [CrossRef]
- Georgiou, M.; Robson, A.G.; Uwaydat, S.H.; Ji, M.H.; Shakarchi, A.F.; Pontikos, N.; Mahroo, O.A.; Cheetham, M.E.; Webster, A.R.; Hardcastle, A.J.; et al. RP2-associated X-linked Retinopathy: Clinical Findings, Molecular Genetics, and Natural History in a Large Cohort of Female Carriers. Am. J. Ophthalmol. 2023, 130, 413–422. [Google Scholar] [CrossRef]
- Duncan, J.L.; Cheng, P.; Maguire, M.G.; Ayala, A.A.; Birch, D.G.; Cheetham, J.K.; Durham, T.A.; Fahim, A.T.; Hoyng, C.B.; Ishikawa, H.; et al. Static Perimetry in the Rate of Progression in USH2A-related Retinal Degeneration (RUSH2A) Study: Assessment Through 2 Years. Am. J. Ophthalmol. 2023, 250, 103–110. [Google Scholar] [CrossRef]
- Birch, D.G.; Samarakoon, L.; Melia, M.; Duncan, J.L.; Ayala, A.R.; Audo, I.; Cheetham, J.K.; Durham, T.A.; Iannaccone, A.; Pennesi, M.E.; et al. The RUSH2A Study: Dark-Adapted Visual Fields in Patients With Retinal Degeneration Associated With Biallelic Variants in the USH2A Gene. Investig. Ophthalmol. Vis. Sci. 2022, 63, 17. [Google Scholar] [CrossRef]
- Lad, E.M.; Duncan, J.L.; Liang, W.; Maguire, M.G.; Ayala, A.R.; Audo, I.; Birch, D.G.; Carroll, J.; Cheetham, J.K.; Durham, T.A.; et al. Baseline Microperimetry and OCT in the RUSH2A Study: Structure-Function Association and Correlation With Disease Severity. Am. J. Ophthalmol. 2022, 244, 98–116. [Google Scholar] [CrossRef]
- Zhu, T.; Chen, D.F.; Wang, L.; Wu, S.; Wei, X.; Li, H.; Jin, Z.B.; Sui, R. USH2A variants in Chinese patients with Usher syndrome type II and non-syndromic retinitis pigmentosa. Br. J. Ophthalmol. 2021, 105, 694–703. [Google Scholar] [CrossRef]
- Pierrache, L.H.; Hartel, B.P.; van Wijk, E.; Meester-Smoor, M.A.; Cremers, F.P.; de Baere, E.; de Zaeytijd, J.; van Schooneveld, M.J.; Cremers, C.W.; Dagnelie, G.; et al. Visual Prognosis in USH2A-Associated Retinitis Pigmentosa Is Worse for Patients with Usher Syndrome Type IIa Than for Those with Nonsyndromic Retinitis Pigmentosa. Ophthalmology 2016, 123, 1151–1160. [Google Scholar] [CrossRef]
- Dad, S.; Rendtorff, N.D.; Kann, E.; Albrechtsen, A.; Mehrjouy, M.M.; Bak, M.; Tommerup, N.; Tranebjærg, L.; Rosenberg, T.; Jensen, H.; et al. Partial USH2A deletions contribute to Usher syndrome in Denmark. Eur. J. Hum. Genet. 2015, 23, 1646–1651. [Google Scholar] [CrossRef]
- Lenassi, E.; Vincent, A.; Li, Z.; Saihan, Z.; Coffey, A.J.; Steele-Stallard, H.B.; Moore, A.T.; Steel, K.P.; Luxon, L.M.; Héon, E.; et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 2015, 23, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Baux, D.; Blanchet, C.; Hamel, C.; Meunier, I.; Larrieu, L.; Faugère, V.; Vaché, C.; Castorina, P.; Puech, B.; Bonneau, D.; et al. Enrichment of LOVD-USHbases with 152 USH2A genotypes defines an extensive mutational spectrum and highlights missense hotspots. Hum. Mutat. 2014, 35, 1179–1186. [Google Scholar] [CrossRef]
- McGee, T.L.; Seyedahmadi, B.J.; Sweeney, M.O.; Dryja, T.P.; Berson, E.L. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J. Med. Genet. 2010, 47, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Auslender, N.; Bandah, D.; Rizel, L.; Behar, D.M.; Shohat, M.; Banin, E.; Allon-Shalev, S.; Sharony, R.; Sharon, D.; Ben-Yosef, T. Four USH2A founder mutations underlie the majority of Usher syndrome type 2 cases among non-Ashkenazi Jews. Genet. Test. 2008, 12, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, B.; Tranebjaerg, L.; Rosenberg, T.; Weston, M.D.; Kimberling, W.J.; Nilssen, O. Identification of novel USH2A mutations: Implications for the structure of USH2A protein. Eur. J. Hum. Genet. 2000, 8, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Weston, M.D.; Eudy, J.D.; Fujita, S.; Yao, S.; Usami, S.; Cremers, C.; Greenberg, J.; Ramesar, R.; Martini, A.; Moller, C.; et al. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am. J. Hum. Genet. 2000, 66, 1199–1210. [Google Scholar] [CrossRef]
- Adato, A.; Weston, M.D.; Berry, A.; Kimberling, W.J.; Bonne-Tamir, A. Three novel mutations and twelve polymorphisms identified in the USH2A gene in Israeli USH2 families. Hum. Mutat. 2000, 15, 388. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.; Villanueva, A.L.; Soto-Hermida, A.; Duncan, J.L.; Matsui, H.; Borooah, S.; Kurmanov, B.; Richard, G.; Khan, S.Y.; Branham, K.; et al. Deciphering the genetic architecture and ethnographic distribution of IRD in three ethnic populations by whole genome sequence analysis. PLoS Genet. 2021, 17, e1009848. [Google Scholar] [CrossRef]
- Kumar, M.; Michael, S.; Alvarado-Valverde, J.; Mészáros, B.; Sámano-Sánchez, H.; Zeke, A.; Dobson, L.; Lazar, T.; Örd, M.; Nagpal, A.; et al. The Eukaryotic Linear Motif resource: 2022 release. Nucleic Acids Res. 2022, 50, D497–D508. [Google Scholar] [CrossRef]
- Nakamura, M.; Hotta, Y.; Tanikawa, A.; Terasaki, H.; Miyake, Y. A high association with cone dystrophy in Fundus albipunctatus caused by mutations of the RDH5 gene. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3925–3932. [Google Scholar] [PubMed]
- Yang, G.; Liu, Z.; Xie, S.; Li, C.; Lv, L.; Zhang, M.; Zhao, J. Genetic and phenotypic characteristics of four Chinese families with fundus albipunctatus. Sci. Rep. 2017, 7, 46285. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Hayashi, T.; Nakamura, M.; Mizobuchi, K.; Gekka, T.; Komori, S.; Ueno, S.; Terasaki, H.; Sakuramoto, H.; Kuniyoshi, K.; et al. RDH5-Related Fundus Albipunctatus in a Large Japanese Cohort. Investig. Ophthalmol. Vis. Sci. 2020, 61, 53. [Google Scholar] [CrossRef]
- Makiyama, Y.; Ooto, S.; Hangai, M.; Ogino, K.; Gotoh, N.; Oishi, A.; Yoshimura, N. Cone abnormalities in fundus albipunctatus associated with RDH5 mutations assessed using adaptive optics scanning laser ophthalmoscopy. Am. J. Ophthalmol. 2014, 157, 558–570.e4. [Google Scholar] [CrossRef]
- Pras, E.; Pras, E.; Reznik-Wolf, H.; Sharon, D.; Raivech, S.; Barkana, Y.; Abu-Horowitz, A.; Ygal, R.; Banin, E. Fundus albipunctatus: Novel mutations and phenotypic description of Israeli patients. Mol. Vis. 2012, 18, 1712–1718. [Google Scholar] [PubMed]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; Sullivan, J.; Williams, S.G.; Lamb, J.A.; Panda, B.; Sergouniotis, P.I.; Gillespie, R.L.; Daiger, S.P.; et al. Molecular findings from 537 individuals with inherited retinal disease. J. Med. Genet. 2016, 53, 761. [Google Scholar] [CrossRef]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Al-Khuzaei, S.; Broadgate, S.; Foster, C.R.; Shah, M.; Yu, J.; Downes, S.M.; Halford, S. An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story. Genes 2021, 12, 1241. [Google Scholar] [CrossRef] [PubMed]
- García Bohórquez, B.; Aller, E.; Rodríguez Muñoz, A.; Jaijo, T.; García García, G.; Millán, J.M. Updating the Genetic Landscape of Inherited Retinal Dystrophies. Front. Cell Dev. Biol. 2021, 9, 645600. [Google Scholar] [CrossRef]
- Chen, E.; Facio, F.M.; Aradhya, K.W.; Rojahn, S.; Hatchell, K.E.; Aguilar, S.; Ouyang, K.; Saitta, S.; Hanson-Kwan, A.K.; Capurro, N.N.; et al. Rates and Classification of Variants of Uncertain Significance in Hereditary Disease Genetic Testing. JAMA Netw. Open 2023, 6, e2339571. [Google Scholar] [CrossRef] [PubMed]
- Landry, L.G.; Rehm, H.L. Association of Racial/Ethnic Categories With the Ability of Genetic Tests to Detect a Cause of Cardiomyopathy. JAMA Cardiol. 2018, 3, 341–345. [Google Scholar] [CrossRef]
- Bea-Mascato, B.; Solarat, C.; Perea-Romero, I.; Jaijo, T.; Blanco-Kelly, F.; Millán, J.M.; Ayuso, C.; Valverde, D. Prevalent ALMS1 Pathogenic Variants in Spanish Alström Patients. Genes 2021, 12, 282. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.D.; Maffei, P.; Collin, G.B.; Naggert, J.K. Alström syndrome: Genetics and clinical overview. Curr. Genom. 2011, 12, 225–235. [Google Scholar] [CrossRef]
- Naeem, M.A.; Chavali, V.R.; Ali, S.; Iqbal, M.; Riazuddin, S.; Khan, S.N.; Husnain, T.; Sieving, P.A.; Ayyagari, R.; Riazuddin, S.; et al. GNAT1 associated with autosomal recessive congenital stationary night blindness. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Méjécase, C.; Stévenard, M.; Michiels, C.; Audo, I.; Marmor, M.F. A Novel Heterozygous Missense Mutation in GNAT1 Leads to Autosomal Dominant Riggs Type of Congenital Stationary Night Blindness. Biomed. Res. Int. 2018, 2018, 7694801. [Google Scholar] [CrossRef]
- Kim, H.M.; Joo, K.; Han, J.; Woo, S.J. Clinical and Genetic Characteristics of Korean Congenital Stationary Night Blindness Patients. Genes 2021, 12, 789. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, M.; Duignan, E.; Humphries, P.; Palfi, A.; Kenna, P.F.; Farrar, G.J. A novel homozygous truncating GNAT1 mutation implicated in retinal degeneration. Br. J. Ophthalmol. 2016, 100, 495–500. [Google Scholar] [CrossRef]
- Zenteno, J.C.; García-Montaño, L.A.; Cruz-Aguilar, M.; Ronquillo, J.; Rodas-Serrano, A.; Aguilar-Castul, L.; Matsui, R.; Vencedor-Meraz, C.I.; Arce-González, R.; Graue-Wiechers, F.; et al. Extensive genic and allelic heterogeneity underlying inherited retinal dystrophies in Mexican patients molecularly analyzed by next-generation sequencing. Mol. Genet. Genom. Med. 2020, 8. [Google Scholar] [CrossRef]
- Calvert, P.D.; Krasnoperova, N.V.; Lyubarsky, A.L.; Isayama, T.; Nicoló, M.; Kosaras, B.; Wong, G.; Gannon, K.S.; Margolskee, R.F.; Sidman, R.L.; et al. Phototransduction in transgenic mice after targeted deletion of the rod transducin α-subunit. Proc. Natl. Acad. Sci. USA 2000, 97, 13913–13918. [Google Scholar] [CrossRef]
- Kubota, D.; Oishi, N.; Gocho, K.; Kikuchi, S.; Yamaki, K.; Igarashi, T.; Takahashi, H.; Ishida, N.; Iwata, T.; Mizota, A.; et al. Novel homozygous in-frame deletion of GNAT1 gene causes golden appearance of fundus and reduced scotopic ERGs similar to that in Oguchi disease in Japanese family. Ophthalmic Genet. 2019, 40, 480–487. [Google Scholar] [CrossRef]
- Van de Sompele, S.; Smith, C.; Karali, M.; Corton, M.; Van Schil, K.; Peelman, F.; Cherry, T.; Rosseel, T.; Verdin, H.; Derolez, J.; et al. Biallelic sequence and structural variants in RAX2 are a novel cause for autosomal recessive inherited retinal disease. Genet. Med. 2019, 21, 1319–1329. [Google Scholar] [CrossRef]
- Simon, A.; Romert, A.; Gustafson, A.L.; McCaffery, J.M.; Eriksson, U. Intracellular localization and membrane topology of 11-cis retinol dehydrogenase in the retinal pigment epithelium suggest a compartmentalized synthesis of 11-cis retinaldehyde. J. Cell Sci. 1999, 112 Pt 4, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Farjo, K.M.; Moiseyev, G.; Takahashi, Y.; Crouch, R.K.; Ma, J.X. The 11-cis-retinol dehydrogenase activity of RDH10 and its interaction with visual cycle proteins. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5089–5097. [Google Scholar] [CrossRef]
- Driessen, C.A.; Winkens, H.J.; Hoffmann, K.; Kuhlmann, L.D.; Janssen, B.P.; Van Vugt, A.H.; Van Hooser, J.P.; Wieringa, B.E.; Deutman, A.F.; Palczewski, K.; et al. Disruption of the 11-cis-retinol dehydrogenase gene leads to accumulation of cis-retinols and cis-retinyl esters. Mol. Cell Biol. 2000, 20, 4275–4287. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Simon, A.; Eriksson, U.; Harris, E.; Berson, E.L.; Dryja, T.P. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat. Genet. 1999, 22, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, M.; Khan, M.I.; Neveling, K.; Khan, Y.M.; Ali, S.H.; Ahmed, W.; Iqbal, M.S.; Azam, M.; den Hollander, A.I.; Collin, R.W.; et al. Novel mutations in RDH5 cause fundus albipunctatus in two consanguineous Pakistani families. Mol. Vis. 2012, 18, 1558–1571. [Google Scholar]
- Occelli, L.M.; Daruwalla, A.; De Silva, S.R.; Winkler, P.A.; Sun, K.; Pasmanter, N.; Minella, A.; Querubin, J.; Lyons, L.A.; Robson, A.G.; et al. A large animal model of RDH5-associated retinopathy recapitulates important features of the human phenotype. Hum. Mol. Genet. 2022, 31, 1263–1277. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Uenoyama, M.; Tomita, N.; Morishita, R.; Kaneda, Y.; Ogihara, T.; Matsumoto, K.; Nakamura, T.; Maruta, A.; Matsuyama, S.; et al. Gene transfer of human hepatocyte growth factor into rat skin wounds mediated by liposomes coated with the sendai virus (hemagglutinating virus of Japan). Am. J. Pathol. 2002, 161, 1761–1772. [Google Scholar] [CrossRef]
- Eisenberger, T.; Neuhaus, C.; Khan, A.O.; Decker, C.; Preising, M.N.; Friedburg, C.; Bieg, A.; Gliem, M.; Charbel Issa, P.; Holz, F.G.; et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: The example of retinal dystrophies. PLoS ONE 2013, 8, e78496. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| ID | Age at Onset/Visit | Race (Self-Reported) | Sex | Inheritance | IRD DX (Syndrome) | Gene | Variant(s) |
|---|---|---|---|---|---|---|---|
| GP2-1 | 6/23 | W | F | R | STGD1 | ABCA4 | c.5461-10T>C/c.179C>T p.(A60V)/c.5603A>T p.(N1868I) |
| GP2-2 | 15/53 | W | F | R | STGD1 | ABCA4 | c.5461-10T>C/c.5056G>A p.(V1686M)/c.5603A>T p.(N1868I) |
| GP2-3 | 26/33 | W | M | R | STGD1 | ABCA4 | c.4253+43G>A; c.2041C>T p.(R681*) |
| GP2-4 | 53/63 | W | F | R | STGD1 | ABCA4 | c.5461-10T>C/c.3113C>T p.(A1038V)/c.5603A>T p.(N1868I) |
| GP2-5 | 54/61 | W | M | R | STGD1 | ABCA4 | c.4685T>C p.(I1562T)/c.4234C>T p.(Q1412*) |
| GP2-6 | 10/35 | W | F | R | CD/CRD | ABCA4 | c.5882G>A p.(G1961E)/c.3113C>T p.(A1038V)/c.1622T>C p.(L541P) |
| GP2-7 | 10/34 | W | F | R | CD/CRD | ABCA4 | c.4539+2028C>T/c.634C>T p.(R212C) |
| GP2-8 | 29/69 | W | F | R | MD | ABCA4 | c.5603A>T p.(N1868I)/c.5734G>A p.(E1912K) ‡ |
| GP2-9 | 25/35 | NA | M | R | STGD1 | ABCA4 | c.6079C>T p.(L2027F)/c.1292G>A p.(W431*) |
| GP2-10 | 32/35 | W | F | R | STGD1 | ABCA4 | c.6317G>A p.(R2106H)/c.5714+5G>A |
| GP2-11 | 32/32 | W | M | R | STGD1 | ABCA4 | c.6079C>T p.(L2027F)/c.768G>T p.(V256=) |
| GP2-12 | 23/31 | W | F | R | STGD1 | ABCA4 | c.6166A>T p.(K2056*)/c.2588G>C p.(G863A)/c.5603A>T p.(N1868I) |
| GP2-41 | 11/11 | W | M | R | RP (Alström) | negative | ALMS1 c.103_108dup p.(A35_A36dup)/c.103_108dup p.(A35_A36dup) ‡ |
| GP2-13 | 9/47 | W | M | R | RP (BBS) | BBS1 | c.1169T>G p.(M390R)/c.1169T>G p.(M390R) |
| GP2-14 | 10/54 | W | F | R | RP (BBS) | BBS1 | c.1169T>G p.(M390R)/c.1169T>G p.(M390R) |
| GP2-15 | 11/37 | W | F | R | RP (BBS) | BBS1 | c.1169T>G p.(M390R)/c.1169T>G p.(M390R) |
| GP2-16 | 12/41 | W | F | R | RP (BBS) | BBS1 | c.1169T>G p.(M390R)/c.800G>T p.(C267F) ‡ |
| GP2-103 | 8/18 | W; A | F | R | RP (BBS) | BBS10 | c.1677C>G p.(Y559*)/c.1121T>C p.(L374P) ‡ |
| GP2-17 | 3/10 | NA | M | R | RP (BBS) | BBS4 | c.332+8T>C/c.1248G>C p.(E416D) § |
| GP2-18 | 4/12 | W | M | D | Best | BEST1 | c.652C>T p.(R218C) |
| GP2-19 | 7/7 | W | M | D | Best | BEST1 | c.878A>G p.(Q293R) |
| GP2-20 | 10/32 | W | M | D | Best | BEST1 | c.275G>A p.(R92H) |
| GP2-21 | 27/28 | W | M | D | Best | BEST1 | c.652C>A p.(R218S) |
| GP2-22 | 8/45 | W | F | R | Best | BEST1 | c.422G>A p.(R141H)/c.602T>C p.(I201T) |
| GP2-23 | 0/81 | H | F | R | RP | CNGB1 | c.2957A>T p.(N986I)/c.2396T>G p.(L799R) § |
| GP2-24 | 3/19 | W | F | R | RP | CRB1 | c.2843G>A p.(C948Y)/c.3014A>T p.(D1005V) § |
| GP2-39 | 63 | W | M | S | MD | negative | CTNNA1 chr5:g.138117546-138473144dup ‡ |
| GP2-44 | 30/64 | W | F | R | RP | negative | DRAM2 c.328G>A p.(A110T)/c.328G>A p.(A110T) ‡ |
| GP2-35 | 6/6 | W | M | S | RP | DYNC2H1 | c.7156G>T p.(G2386*)/c.10205_10207del p.(T3402del) ‡ |
| GP2-25 | 28/74 | W | M | R | RP | EYS | c.7095T>G p.(Y2365*)/c.1505_1506insGA p.(F502Lfs*14) § |
| GP2-104 | 20/24 | W | M | R | RP | EYS | c.2528G>A p.(G843E)/c.(3443+1_3444-1)_(6424+1_6425-1)del encompassing exons 23–31 |
| GP2-26 | 20/21 | H | F | D | FEVR | FZD4 | c.313A>G p.(M105V) |
| GP2-27 | 0/68 | H | M | R | RP | GNAT1 | c.758_771delinsC p.(R253Pfs*56) |
| GP2-34 | 1/41 | B | M | D | CD/CRD | GUCY2D | c.2578T>C p.(S860P) ‡ |
| GP2-107 | 5/28 | W | M | D | RP | IMPDH1 | c.931G>A p.(D311N) |
| GP2-28 | 10/41 | W | F | S | FEVR | KIF11 | c.308+1G>A |
| GP2-29 | 16/33 | W | F | MT | RP | negative | MT-ND4 m.11778G>A (100%) |
| GP2-30 | 42/45 | W | F | MT | PD (MMSD) | MT-TL1 | m.3243A>G (15.3%) |
| GP2-31 | 60/61 | W | F | MT | MD (MSMD) | MT-TL1 | m.3243A>G (28.8%) |
| GP2-32 | 21/49 | W | F | MT | RP (MSMD) | MT-TL1 | m.3255G>A (24.8%) |
| GP2-33 | 0/57 | W | F | R | RP (USH1) | MYO7A | c.1559_1560del p.(T520Rfs*26)/c.5302C>T p.(Q1768*) |
| GP2-37 | 13/13 | NA | F | S | MD | negative | |
| GP2-40 | 10/61 | NA | F | D or XL | MD | negative | |
| GP2-43 | 25/36 | NA | F | S | RP | negative | |
| GP2-45 | 32/40 | W | M | S | RP | negative | OPA1 c.2708_2711del p.(V903Gfs*3) |
| GP2-46 | 50/60 | W | F | S | RP (Stickler) | negative | |
| GP2-47 | 60/60 | H | F | S | RP | negative | |
| GP2-48 | 62/68 | A | F | S | RP | negative | |
| GP2-49 | 12/32 | H | F | S | RP (USH2) | negative | |
| GP2-50 | 31/43 | B | M | S | RP | negative | |
| GP2-51 | 50/58 | W | F | S | RP | negative | |
| GP2-52 | 9/20 | W | M | S | MD | negative | |
| GP2-53 | 61/61 | NA | M | S | MD | negative | |
| GP2-54 | 37/43 | W | F | S | MD | negative | |
| GP2-55 | 28/39 | NA | M | D or XL | RP | negative | |
| GP2-56 | 16/54 | W | F | S | RP | negative | |
| GP2-57 | 25/54 | A | F | S | RP | negative | |
| GP2-58 | 30/67 | NA | M | S | RP | negative | |
| GP2-59 | 35/75 | W | F | S | RP | negative | |
| GP2-60 | 40/65 | W | M | S | RP | negative | |
| GP2-61 | 45/56 | H | M | D or XL | RP | negative | |
| GP2-62 | 48/55 | NA | F | S | RP | negative | |
| GP2-63 | 51/55 | W | F | S | RP | negative | |
| GP2-64 | 5/24 | W | M | S | RP | negative | |
| GP2-65 | 46/66 | W | M | S | STGD1 | negative | |
| GP2-66 | 59/78 | W | F | R | STGD1 | negative | ABCA4 c.5603A>T p.(N1868I) |
| GP2-67 | 59/63 | W | M | S | XLRS | negative | |
| GP2-100 | 30/60 | H | M | S | RP | negative | |
| GP2-101 | 34/40 | W | F | S | MD | negative | |
| GP2-106 | 0/36 | B | M | S | MD | negative | |
| GP2-68 | 6/8 | NA | F | R | MD | NMNAT1 | c.507G>A p.(W169*)/c.709C>T p.(R237C) |
| GP2-69 | 8/47 | W | F | R | RP | NR2E3 | c.119-2A>C; c.767C>A p.(A256E) |
| GP2-70 | 13/21 | B | M | D | MD | PROM1 | c.1117C>T p.(R373C) |
| GP2-71 | 6/39 | W | M | D | RP | PRPF31 | c.(238+1_239-1)_(1374+1_1375-1)dup exons 4–13 |
| GP2-72 | 25/60 | W | M | D | RP | PRPF31 | c.697+1G>C |
| GP2-73 | 38/48 | W | F | D | RP | PRPF31 | c.1031_1032del p.(P344Rfs*130) |
| GP2-74 | 9/71 | W | M | D | MD | PRPH2 | c.907_910delinsCG p.(S303Rfs*88) |
| GP2-75 | 40/60 | NA | M | D | MD | PRPH2 | c.629C>G p.(P210R) |
| GP2-76 | 40/43 | B | M | D | MD | PRPH2 | c.515G>A p.(R172Q) |
| GP2-77 | 45/63 | W | M | D | MD | PRPH2 | c.828+3A>T p.(?) |
| GP2-78 | 53/71 | W | M | D | PD | PRPH2 | c.(828+1_829-1)_(1*_?)del |
| GP2-79 | 24/56 | W | M | D | RP | PRPH2 | c.828+3A>T, p(?) |
| GP2-80 | 53/82 | W | F | D | MD | PRPH2 | c.620A>G p.(D207G) |
| GP2-81 | 20/51 | W | M | R | RP | RAX2 | c.236G>A p.(R79Q)/c.181G>A p.(A61T) ‡ |
| GP2-82 | 7/7 | H | M | R | FA | RDH5 | c.718dup p.(A240Gfs*19)/c.833G>A p.(R278Q) § |
| GP2-83 | 11/33 | NA | F | D | RP | RHO | c.509C>G p.(P170R) |
| GP2-84 | 13/34 | W | F | D | RP | RHO | c.152G>T p.(G51V) |
| GP2-85 | 10/45 | A | M | D | RP | RHO | c.404_405delinsTT p.(R135L) |
| GP2-86 | 39/42 | NA | F | S | RP | RP2 | c.969G>C p.(K323N) ‡ |
| GP2-87 | 7/7 | A | F | R | CD/CRD | RPE65 | c.1102T>C p.(Y368H)/c.718G>T p.(V240F) |
| GP2-88 | 10/30 | H | F | R | RP | RPE65 | c.1067dup p.(N356Kfs*9)/c.1067dup p.(N356Kfs*9) |
| GP2-89 | 0/61 | W | M | XL | MD | RPGR | c.3178_3179del p.(E1060Rfs*18) |
| GP2-90 | 3/5 | H | F | XL | RP | RPGR | c.2362G>T p.(E788*) |
| GP2-91 | 12/23 | NA | M | XL | RP | RPGR | c.2146G>T p.(E716*) |
| GP2-92 | 5/35 | H | M | XL | XLRS | RS1 | c.208G>A p.(G70S) |
| GP2-93 | 20/20 | W | M | XL | XLRS | RS1 | CNV c.(52+1_53-1)_(78+1_79-1)del |
| GP2-94 | 16/73 | W | M | R | RP | USH2A | c.12295-2A>G/c.10073G>A p.(C3358Y) |
| GP2-95 | 26/28 | H | M | R | RP (USH2) | USH2A | c.1000C>T p.(R334W)/c.1000C>T p.(R334W) |
| GP2-96 | 54/55 | B | M | R | RP | USH2A | c.12145G>A p.(A4049T)/c.274T>G p.(S92A) ‡ |
| GP2-97 | 50/72 | NA | M | R | RP | USH2A | c.2384G>A p.(C795Y)/c.11712-3520_11712-3519ins[GTAAGGATTCCCAATAATCCTTACCCT] |
| GP2-98 | 30/46 | W | M | R | RP (USH2) | USH2A | c.(5298+1_5299-1) (5572+1_5573-1)del/c.14851T>G p.(W4951G) ‡, c.253A>T p.(I85F) § |
| GP2-99 | 2/40 | W | F | R | RP (USH2) | USH2A | c.13374del p.(E4458Dfs*3)/c.12152_12153insTT p.(E4051Dfs*2) |
| GP2-105 | 17/33 | B | M | R | RP (USH2) | USH2A | c.6428del p.(P2143Qfs*9)/c.4001A>G p.(K1334R) ‡ |
| Gene (Inheritance) | Variant | gnomAD Allele Frequency | PolyPhen | SIFT | MUT-TASTER | Mut-Pred2 | MutPred2 Mechanisms p < 0.05 | ACMG Criteria |
|---|---|---|---|---|---|---|---|---|
| ALMS1 (Rhom) | c.103_108dup p.(A35_A36dup) | 0.0005103 | N/A | N/A | N/A | 0.29022 | Iron binding | PP4, BP3 |
| BBS1 (Rhet) | c.800G>T p.(C267F) | 0 | PrD | D | DC | 0.925 | Altered DNA binding, altered transmembrane protein, loss of disulfide linkage at C267 | PM2, PP3, PP4 |
| BBS4 (Rhet) | c.1248G>C p.(E416D) § | 0 | PoD | T | DC | 0.411 | None | PM2, PP3, PP4, PM3 |
| BBS10 (Rhet) | c.1677C>G p.(Y559*) | 0 | PrD | D | DC | 0.52879 | N/A | PM2, PVS1, PP4 |
| BBS10 (Rhet) | c.1121T>C p.(L374P) | 0 | PrD | D | DC | 0.484 | None | PM2, PP4 |
| CTNNA1 (d) | chr5:g.138117546-138473144dup | 0 | N/A | N/A | N/A | 0.37257 | N/A | PM2 |
| DRAM2 (Rhom) | c.328G>A p.(A110T) | 3.188 × 10−5 | PrD | D | DC | 0.744 | Loss of helix, gain of strand, altered transmembrane protein | PP3 |
| DYNC2H1 (Rhet) | c.10205_10207del p.(T3402del) | 0.000125 | N/A | N/A | N/A | 0.45459 | Iron-binding; catalytic site | PM2 |
| GNAT1 (Rhom) | c.758_771delinsC p.(Arg253Pfs*56) | 0 | N/A | N/A | N/A | 0.40539 | N/A | PM2, PM4 |
| GUCY2D (d) | c.2578T>C p.(S860P) | 3.978 × 10−6 | PrD | D | DC | 0.597 | Altered coiled-coil, altered transmembrane protein | PP3 |
| PRPH2 (d) | c.907_910delinsCG p.(S303Rfs*88) | 0 | N/A | N/A | N/A | 0.31587 | N/A | PM2, PM4 |
| PRPH2 (d) | c.620A>G p.(D207G) | 0 | PrD | D | DC | 0.836 | Altered transmembrane protein, loss of proteolytic cleavage at R203 | PM2, PP3 |
| RAX2 (Rhet) | c.181G>A p.(A61T) | 8.326 × 10−6 | PrD | D | DC | 0.618 | Gain of allosteric site at R57, metal-binding | PP3 |
| RDH5 (Rhet) | c.833G>A p.(R278Q) § | 2.386 × 10−5 | PrD | D | DC | 0.659 | Altered metal-binding, gain of catalytic site at Y281 | PM3, PP3 |
| RHO (d) | c.404_405delinsTT, p.(Arg135Leu) | 0 | PrD | D | DC | 0.941 | Loss of strand, gain of helix, loss of disulfide linkage at C140, altered ordered interface, altered transmembrane protein | PM2, PP3 |
| RP2 (x) | c.969G>C p.(K323N) | 0 | B | T | DC | 0.43 | None | PM2, PP3, BP4 |
| USH2A (Rhet) | c.274T>G p.(S92A) | 7.971 × 10−6 | B | D | DC | 0.216 | None | PP4, PP3, PP4 |
| USH2A (Rhet) | c.253A>T p.(I85F) § | 0 | PrD | D | DC | 0.328 | None | PM2, PP3, PM3 |
| USH2A (Rhet) | c.4001A>G p.(K1334R) | 7.081 × 10−6 | B | T | PM | 0.105 | None | PP4, BP4 |
| USH2A (Rhet) | c.2384G>A p.(C795Y) | 0 | PrD | D | DC | 0.921 | Altered transmembrane protein, gain of disulfide linkage at C792, altered metal binding, gain of catalytic site at C795, gain of sulfation at C795 | PM2, PP3, PP4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lynn, J.; Huang, S.J.; Trigler, G.K.; Kingsley, R.; Coussa, R.G.; Bennett, L.D. Expanding the Mutation Spectrum for Inherited Retinal Diseases. Genes 2025, 16, 32. https://doi.org/10.3390/genes16010032
Lynn J, Huang SJ, Trigler GK, Kingsley R, Coussa RG, Bennett LD. Expanding the Mutation Spectrum for Inherited Retinal Diseases. Genes. 2025; 16(1):32. https://doi.org/10.3390/genes16010032
Chicago/Turabian StyleLynn, Jacob, Samuel J. Huang, Grace K. Trigler, Ronald Kingsley, Razek G. Coussa, and Lea D. Bennett. 2025. "Expanding the Mutation Spectrum for Inherited Retinal Diseases" Genes 16, no. 1: 32. https://doi.org/10.3390/genes16010032
APA StyleLynn, J., Huang, S. J., Trigler, G. K., Kingsley, R., Coussa, R. G., & Bennett, L. D. (2025). Expanding the Mutation Spectrum for Inherited Retinal Diseases. Genes, 16(1), 32. https://doi.org/10.3390/genes16010032