
Complete Mitochondrial Genome of King Threadfin, Polydactylus macrochir (Günther, 1867): Genome Characterization and Phylogenetic Analysis

Abstract
1. Introduction
2. Materials and Methods
2.1. Samples, DNA Extraction and Sequencing
2.2. Genome Assembly and Annotation
2.3. Codon Usage Analysis
2.4. Ka/Ks Value Analyses
2.5. Phylogenetic Analysis TimeTree Estimation
3. Results
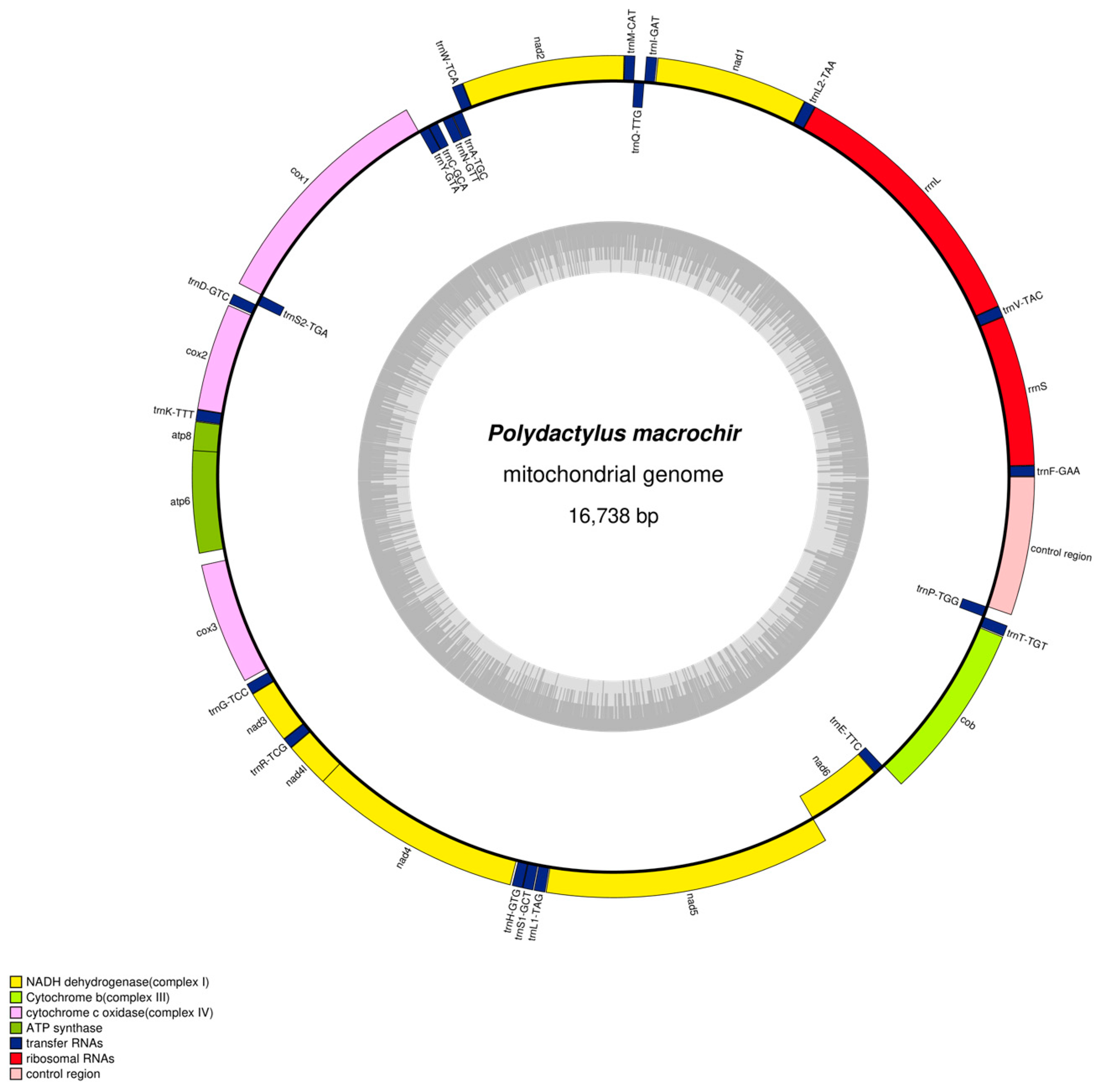
3.1. Mitogenomic Structure and Organization
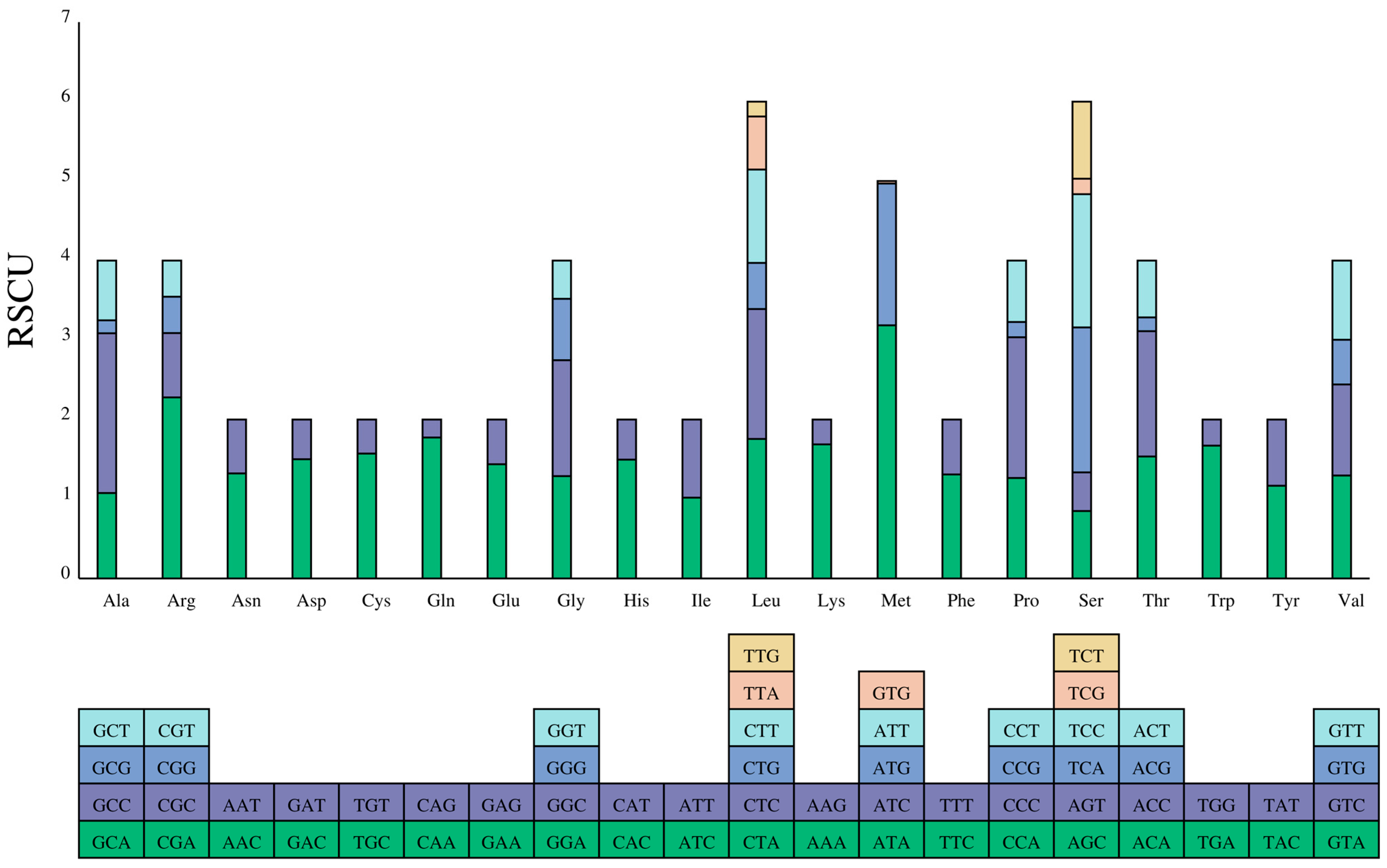
3.2. Protein-Coding Genes and Codon Usage
3.3. Ka/Ks Value Analyses
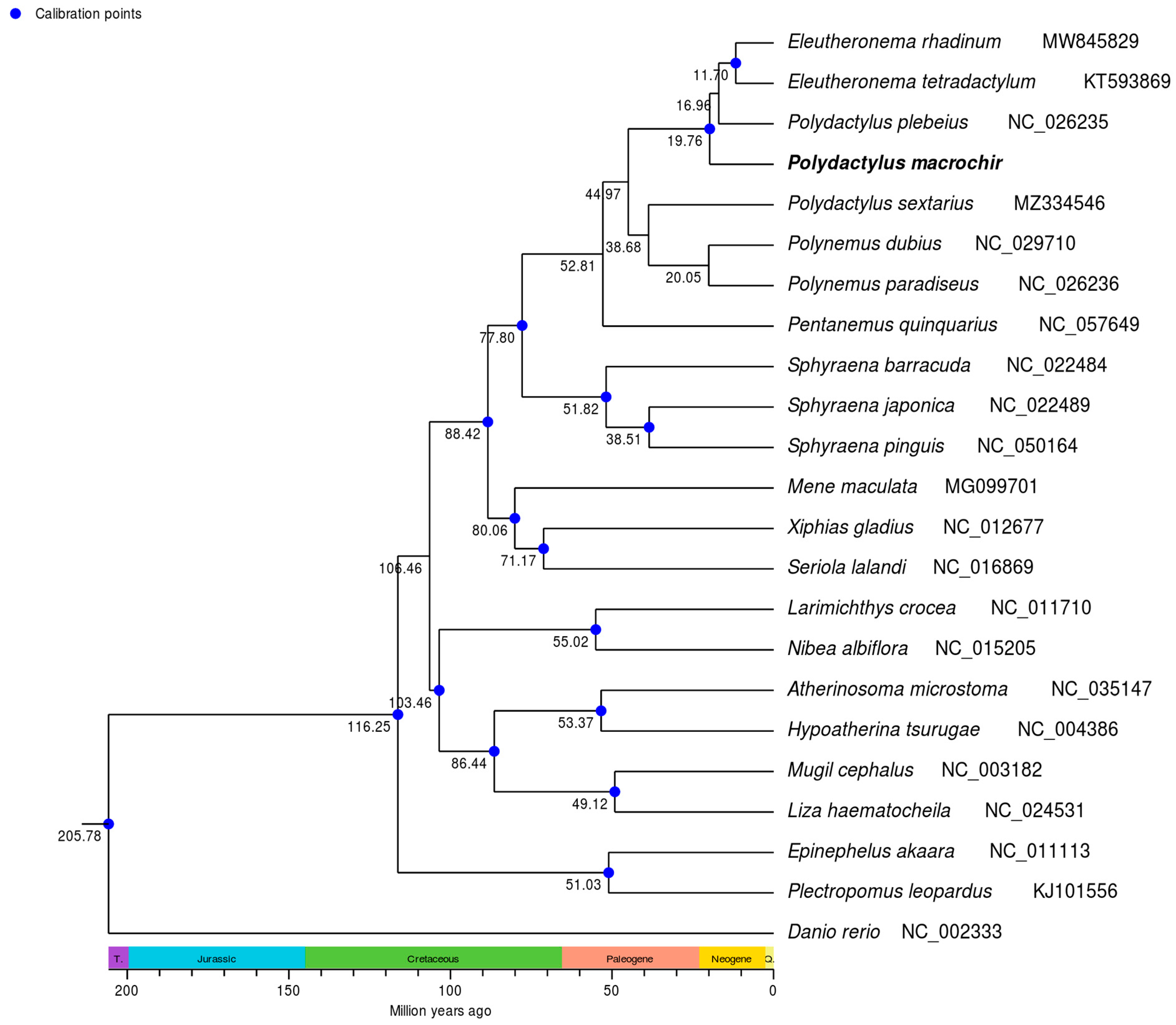
3.4. Phylogenetic Analysis and TimeTree Estimation
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Motomura, H. Treadfns of the World (Family Polynemidae): An Annotated and Illustrated Catalogue of Polynemid Species Known to Date; Food & Agriculture Organization: Rome, Italy, 2004. [Google Scholar]
- Presti, P.; Johnson, G.D.; Datovo, A. Anatomy and evolution of the pectoral filaments of threadfins (Polynemidae). Sci. Rep. 2020, 10, 17751. [Google Scholar] [CrossRef]
- Fricke, R.; Eschmeyer, W.; Fong, J. Eschmeyer’s Catalog of Fishes: Genera/Species by Family/Subfamily; California Academy of Sciences: San Francisco, CA, USA, 2022. [Google Scholar]
- Ou, Y.J.; Xie, M.J.; Li, J.E.; Wen, J.F.; Zhou, H.; Liu, Q.Q. First maturation and mass seedling propagation of cultured Eleutheronema tetradactylum in Guangdong Province. S. China Fish. Sci. 2017, 13, 97–104. (In Chinese) [Google Scholar]
- Niu, Y.Y.; Ou, Y.J.; Lan, J.N.; Wen, J.F.; Li, J.E.; Li, J.W.; Zhou, H. Structure and early development of gill tissue in artificially cultured Eleutheronema tetradactylum. S. China Fish. Sci. 2020, 16, 108–114. (In Chinese) [Google Scholar]
- Gosline, W.A. Systematic position and relationships of the percesocine fishes. Pac. Sci. 1962, 16, 207–217. [Google Scholar]
- Rosen, D.E. The relationships and taxonomic position of the halfbeaks, killifishes, silversides, and their relatives. Bull. Am. Mus. Nat. Hist. 1964, 127, 217–268. [Google Scholar]
- Johnson, D.G. Percomorph phylogeny: Progress and problems. Bull. Mar. Sci. 1993, 52, 3–28. [Google Scholar]
- Kang, S.; Imamura, H.; Kawai, T. Morphological evidence supporting the monophyly of the family Polynemidae (Teleostei: Perciformes) and its sister relationship with Sciaenidae. Ichthyol. Res. 2017, 65, 29–41. [Google Scholar] [CrossRef]
- Presti, P.; Johnson, G.D.; Datovo, A. Facial and gill musculature of polynemid fishes, with notes on their possible relationships with sciaenids (Percomorphacea: Perciformes). J. Morphol. 2020, 281, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Mirande, J.M. Combined phylogeny of ray-ffnned fishes (Actinopterygii) and the use of morphological characters in large-scale analyses. Cladistics 2017, 33, 333–350. [Google Scholar] [CrossRef]
- Sanciangco, M.D.; Carpenter, K.E.; Betancur-R, R. Phylogenetic placement of enigmatic percomorph families (Teleostei: Percomorphaceae). Mol. Phylogenetics Evol. 2015, 94, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Betancur-R, R.; Broughton, R.E.; Wiley, E.O.; Carpenter, K.; López, J.A.; Li, C.; Holcroft, N.I.; Arcila, D.; Sanciangco, M.; Cureton Ii, J.C.; et al. The tree of life and a new classification of bony fishes. PLoS Curr. 2013, 5, 23653398. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.C.; Ortí, G.; Huang, Y.; Sun, Y.; Baldwin, C.C.; Thompson, A.W.; Arcila, D.; Betancur-R, R.; Li, C.; Becker, L.; et al. Comprehensive phylogeny of rayffnned ffshes (Actinopterygii) based on transcriptomic and genomic data. Proc. Natl. Acad. Sci. USA 2018, 115, 6249–6254. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.G.; Davis, M.P.; Smith, W.L. The phylogeny of Carangiform fishes: Morphological and genomic investigations of a new fish clade. Copeia 2020, 108, 265–298. [Google Scholar] [CrossRef]
- Zhong, L.Q.; Wang, M.H.; Li, D.M.; Tang, S.K.; Chen, X.H. Mitochondrial genome of Eleutheronema rhadinum with an additional non-coding region and novel insights into the phylogenetics. Front. Mar. Sci. 2021, 8, 746598. [Google Scholar] [CrossRef]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Moritz, C.; Dowling, T.E.; Brown, W.M. Evolution of animal mitochondrial DNA: Relevance for population biology and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 269–292. [Google Scholar] [CrossRef]
- Wang, C.; Ye, P.; Liu, M.; Zhang, Y.; Feng, H.; Liu, J.; Zhou, H.; Wang, J.; Chen, X. Comparative Analysis of Four Complete Mitochondrial Genomes of Epinephelidae (Perciformes). Genes 2022, 13, 660. [Google Scholar] [CrossRef]
- Gao, J.; Li, C.; Yu, D.; Wang, T.; Lin, L.; Xiao, Y.; Wu, P.; Liu, Y. Comparative mitogenome analyses uncover mitogenome features and phylogenetic implications of the parrotfishes (Perciformes: Scaridae). Biology 2023, 12, 410. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.M.; Xue, J.F.; Zhao, X.M.; Ding, K.; Liu, Z.W.; Wang, Z.S.; Chen, J.B.; Huang, Y.K. Mitochondrial genome characteristics reveal evolution of Acanthopsetta nadeshnyi (Jordan and Starks, 1904) and phylogenetic relationships. Genes 2024, 15, 893. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A Toolkit incorporating γ-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Montaña-Lozano, P.; Moreno-Carmona, M.; Ochoa-Capera, M.; Medina, N.S.; Boore, J.L.; Prada, C.F. Comparative genomic analysis of vertebrate mitochondrial reveals a differential of rearrangements rate between taxonomic class. Sci. Rep. 2022, 12, 5479. [Google Scholar] [CrossRef]
- Ke, Z.; Zhou, K.; Hou, M.; Luo, H.; Li, Z.; Pan, X.; Zhou, J.; Jing, T.; Ye, H. Characterization of the Complete Mitochondrial Genome of the Elongate Loach and Its Phylogenetic Implications in Cobitidae. Animals 2023, 13, 3841. [Google Scholar] [CrossRef] [PubMed]
- Gun, L.; Yumiao, R.; Haixian, P.; Liang, Z. Comprehensive analysis and comparison on the codon usage pattern of whole Mycobacterium tuberculosis coding genome from different area. Biomed. Res. Int. 2018, 2018, 3574976. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Kang, H.E.; Kim, A.R.; Lee, S.R.; Kim, E.B.; Amin, M.H.F.; Andriyono, S.; Kim, H.W.; Kang, K. Mitogenomic Characterization and Phylogenetic Placement of African Hind, Cephalopholis taeniops: Shedding Light on the Evolution of Groupers (Serranidae: Epinephelinae). Int. J. Mol. Sci. 2024, 25, 1822. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Keating, J.N.; Garwood, R.J.; Sansom, R.S. Phylogenetic congruence, conflict and consilience between molecular and morphological data. BMC Ecol. Evol. 2023, 23, 30. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Family | Genus | Species | Size (bp) | Accession No. |
|---|---|---|---|---|---|
| Perciformes | Polynemidae | Eleutheronema | Eleutheronema rhadinum | 16,718 | MW845829.1 |
| Eleutheronema tetradactylum | 16,491 | KT593869.1 | |||
| Pentanemus | Pentanemus quinquarius | 16,708 | NC_057649.1 | ||
| Polydactylus | Polydactylus sextarius | 16,841 | MZ334546.1 | ||
| Polydactylus plebeius | 16,765 | NC_026235.1 | |||
| Polynemus | Polynemus paradiseus | 16,710 | NC_026236.1 | ||
| Polynemus dubius | 16,555 | NC_029710.1 | |||
| Sphyraenidae | Sphyraena | Sphyraena pinguis | 16,620 | NC_050164.1 | |
| Sphyraena barracuda | 16,707 | NC_022484.1 | |||
| Sphyraena japonica | 16,760 | NC_022489.1 | |||
| Xiphiidae | Xiphias | Xiphias gladius | 16,520 | NC_012677.1 | |
| Carangidae | Seriola | Seriola lalandi | 16,532 | NC_016869.1 | |
| Menidae | Mene | Mene maculata | 16,733 | MG099701.1 | |
| Sciaenidae | Larimichthys | Larimichthys crocea | 16,466 | NC_011710.1 | |
| Nibea | Nibea albiflora | 16,499 | NC_015205.1 | ||
| Atheriniformes | Atherinidae | Atherinosoma | Atherinosoma microstoma | 16,573 | NC_035147.1 |
| Hypoatherina | Hypoatherina tsurugae | 16,566 | NC_004386.1 | ||
| Serranidae | Epinephelus | Epinephelus akaara | 16,795 | NC_011113.1 | |
| Plectropomus | Plectropomus leopardus | 16,754 | KJ101556.1 | ||
| Mugiliformes | Mugilidae | Mugil | Mugil cephalus | 16,685 | NC_003182.1 |
| Planiliza | Liza haematocheila | 16,822 | NC_024531.1 | ||
| Cypriniformes | Danionidae | Danio | Danio rerio | 16,596 | NC_002333.2 |
| Position | Codon | ||||||
|---|---|---|---|---|---|---|---|
| Gene | Stand | From | To | Size | Intergenic Length | Start | Stop |
| trnF | H | 1 | 70 | 70 | 0 | ||
| rrnS | H | 71 | 1037 | 967 | 0 | ||
| trnV | H | 1038 | 1110 | 73 | 0 | ||
| rrnL | H | 1111 | 2853 | 1743 | 0 | ||
| trnL2 | H | 2854 | 2927 | 74 | 0 | ||
| nad1 | H | 2928 | 3902 | 975 | 0 | ATG | TAA |
| trnI | H | 3907 | 3976 | 70 | 4 | ||
| trnQ | L | 3976 | 4046 | 71 | −1 | ||
| trnM | H | 4046 | 4114 | 69 | −1 | ||
| nad2 | H | 4115 | 5160 | 1046 | 0 | ATG | TA- |
| trnW | H | 5161 | 5231 | 71 | 0 | ||
| trnA | L | 5233 | 5301 | 69 | 1 | ||
| trnN | L | 5303 | 5375 | 73 | 1 | ||
| trnC | L | 5414 | 5479 | 66 | 38 | ||
| trnY | L | 5480 | 5549 | 70 | 0 | ||
| cox1 | H | 5551 | 7101 | 1551 | 1 | GTG | TAA |
| trnS2 | L | 7102 | 7173 | 72 | 0 | ||
| trnD | H | 7174 | 7242 | 69 | 0 | ||
| cox2 | H | 7252 | 7942 | 691 | 9 | ATG | T-- |
| trnK | H | 7943 | 8016 | 74 | 0 | ||
| atp8 | H | 8018 | 8206 | 189 | 1 | ATG | TAA |
| atp6 | H | 8176 | 8859 | 684 | −31 | ATG | TAA |
| cox3 | H | 8933 | 9718 | 786 | 73 | ATG | TAA |
| trnG | H | 9742 | 9813 | 72 | 23 | ||
| nad3 | H | 9814 | 10,162 | 349 | 0 | ATG | T-- |
| trnR | H | 10,163 | 10,231 | 69 | 0 | ||
| nad4l | H | 10,232 | 10,528 | 297 | 0 | ATG | TAA |
| nad4 | H | 10,522 | 11,896 | 1375 | −7 | ATG | T-- |
| trnH | H | 11,906 | 11,974 | 69 | 9 | ||
| trnS1 | H | 11,975 | 12,043 | 69 | 0 | ||
| trnL1 | H | 12,048 | 12,120 | 73 | 4 | ||
| nad5 | H | 12,124 | 13,962 | 1839 | 3 | ATG | TAA |
| nad6 | L | 13,959 | 14,480 | 522 | −4 | ATG | TAG |
| trnE | L | 14,482 | 14,551 | 70 | 1 | ||
| cob | H | 14,556 | 15698 | 1143 | 4 | ATG | TAA |
| trnT | H | 15,703 | 15,774 | 72 | 4 | ||
| trnP | L | 15,774 | 15,845 | 72 | −1 | ||
| D-loop | H | 15,846 | 16,738 | 893 | 0 | ||
| Polydactylus_Macrochir | Size(bp) | A% | T% | G% | C% | A + T% | G + C% | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|---|
| Mitogenome | 16,738 | 28.4 | 25.75 | 16.36 | 29.49 | 54.15 | 45.85 | 0.049 | −0.286 |
| PCGs | 11,447 | 25.6 | 27.71 | 15.65 | 31.03 | 53.32 | 46.68 | −0.039 | −0.329 |
| tRNAs | 1557 | 27.49 | 26.59 | 23.83 | 22.09 | 54.08 | 45.92 | 0.017 | 0.038 |
| rRNAs | 2710 | 31.51 | 22.77 | 20.92 | 24.8 | 54.28 | 45.72 | 0.161 | −0.085 |
| Dloop | 893 | 35.72 | 31.02 | 14.22 | 19.04 | 66.74 | 33.26 | 0.07 | −0.145 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, J. Complete Mitochondrial Genome of King Threadfin, Polydactylus macrochir (Günther, 1867): Genome Characterization and Phylogenetic Analysis. Genes 2025, 16, 88. https://doi.org/10.3390/genes16010088
Wen J. Complete Mitochondrial Genome of King Threadfin, Polydactylus macrochir (Günther, 1867): Genome Characterization and Phylogenetic Analysis. Genes. 2025; 16(1):88. https://doi.org/10.3390/genes16010088
Chicago/Turabian StyleWen, Jiufu. 2025. "Complete Mitochondrial Genome of King Threadfin, Polydactylus macrochir (Günther, 1867): Genome Characterization and Phylogenetic Analysis" Genes 16, no. 1: 88. https://doi.org/10.3390/genes16010088
APA StyleWen, J. (2025). Complete Mitochondrial Genome of King Threadfin, Polydactylus macrochir (Günther, 1867): Genome Characterization and Phylogenetic Analysis. Genes, 16(1), 88. https://doi.org/10.3390/genes16010088