
Genotype–Phenotype Correlations of Nance–Horan Syndrome in Male and Female Carriers of a Novel Variant

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Clinical Evaluation
2.3. Genomic DNA
2.4. Library Preparation and Whole-Exome Sequencing
2.5. Bioinformatics Analysis
2.6. Sanger Sequencing
3. Results
3.1. Ophthalmological Exam
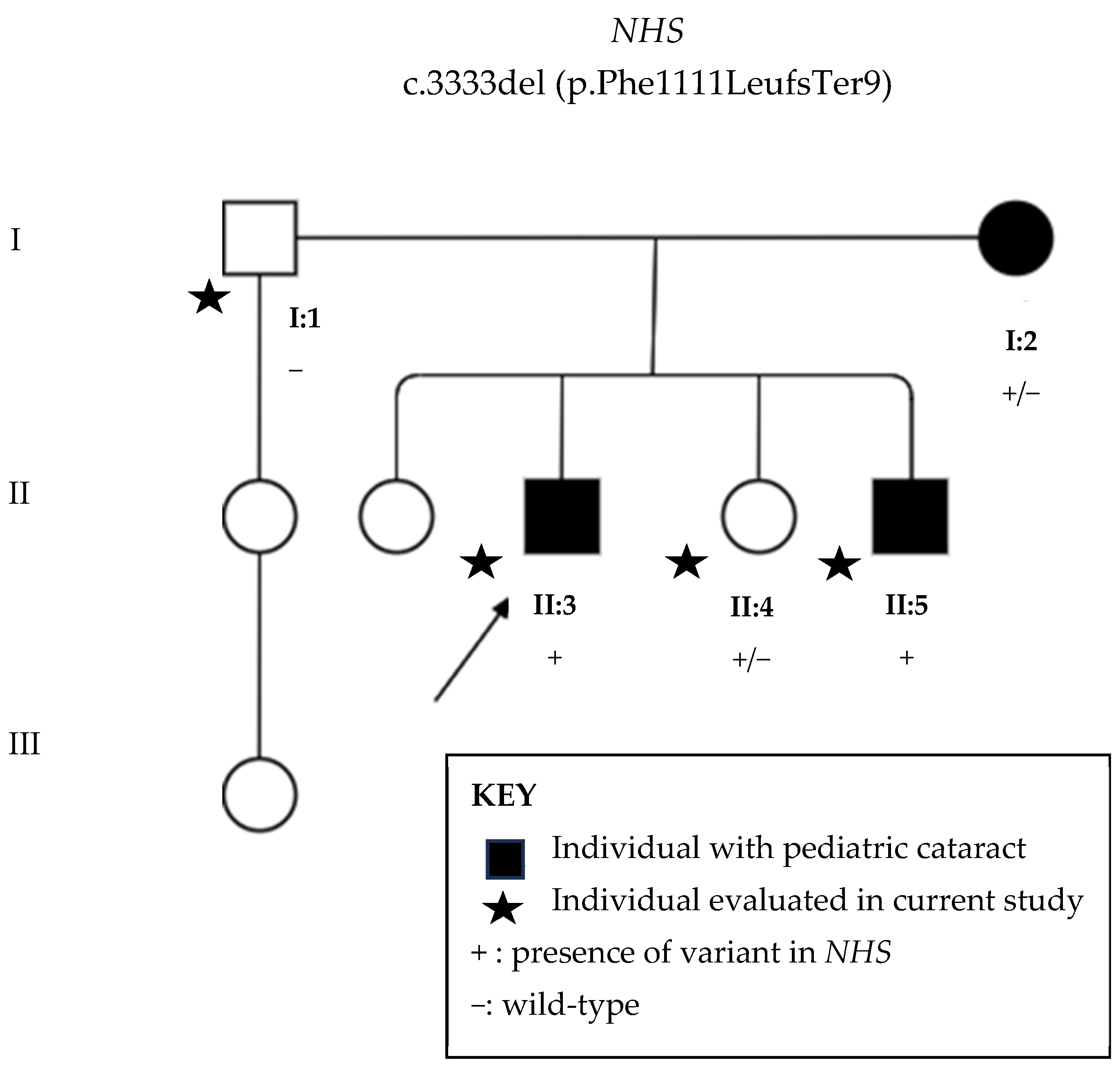
3.2. Next-Generation Sequencing
3.3. Sanger Sequencing
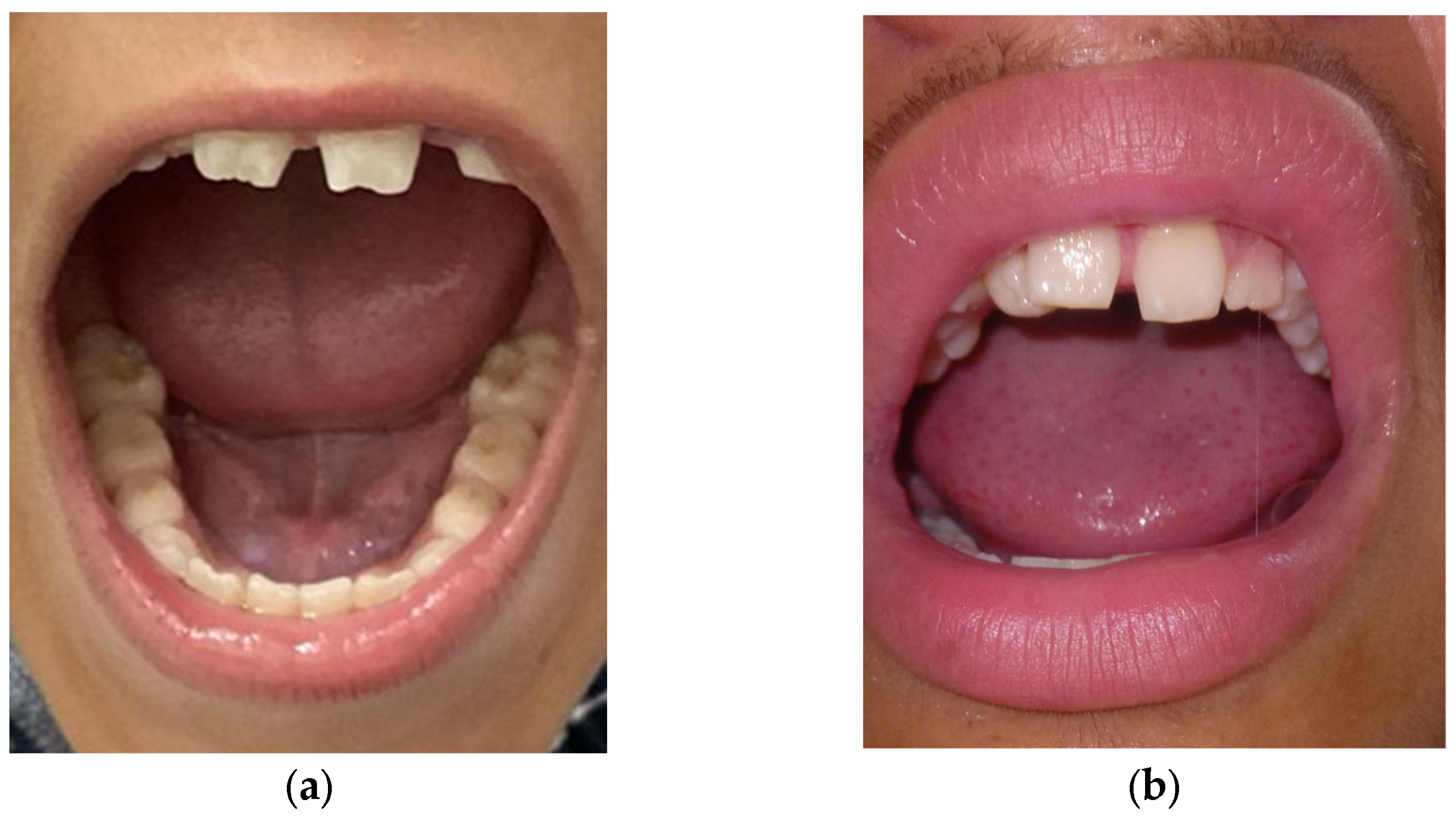
3.4. Systemic Findings
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walpole, I.R.; Hockey, A.; Nicoll, A. The Nance-Horan Syndrome. J. Med. Genet. 1990, 27, 632–634. [Google Scholar] [CrossRef] [PubMed]
- Guven, Y.; Saracoglu, H.P.; Aksakal, S.D.; Kalayci, T.; Altunoglu, U.; Uyguner, Z.O.; Eraslan, S.; Borklu, E.; Kayserili, H. Nance-Horan Syndrome: Characterization of dental, clinical and molecular features in three new families. BMC Oral. Health 2023, 23, 314. [Google Scholar] [CrossRef] [PubMed]
- Coccia, M.; Brooks, S.P.; Webb, T.R.; Christodoulou, K.; Wozniak, I.O.; Murday, V.; Balicki, M.; Yee, H.A.; Wangensteen, T.; Riise, R.; et al. X-linked cataract and Nance-Horan syndrome are allelic disorders. Hum. Mol. Genet. 2009, 18, 2643–2655. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ma, L.; Zhang, Z.; Nie, S.; Zhou, Y.; Zhang, J.; Wang, C.; Fang, X.; Quan, Y.; He, T.; et al. Nance-Horan syndrome pedigree due to a novel microdeletion and skewed X chromosome inactivation. Mol. Genet. Genom. Med. 2023, 11, e2100. [Google Scholar] [CrossRef] [PubMed]
- Damián, A.; Ionescu, R.O.; Rodríguez de Alba, M.; Tamayo, A.; Trujillo-Tiebas, M.J.; Cotarelo-Pérez, M.C.; Pérez Rodríguez, O.; Villaverde, C.; de la Fuente, L.; Romero, R.; et al. Fine Breakpoint Mapping by Genome Sequencing Reveals the First Large X Inversion Disrupting the NHS Gene in a Patient with Syndromic Cataracts. Int. J. Mol. Sci. 2021, 22, 12713. [Google Scholar] [CrossRef]
- Gómez-Laguna, L.; Martínez-Herrera, A.; de la Rosa, A.P.R.; García-Delgado, C.; Nieto-Martínez, K.; Fernández-Ramírez, F.; Valderrama-Atayupanqui, T.Y.; Morales-Jiménez, A.B.; Villa-Morales, J.; Kofman, S.; et al. Nance–Horan syndrome in females due to a balanced X;1 translocation that disrupts the NHS gene: Familial case report and review of the literature. Ophthalmic Genet. 2017, 39, 56–62. [Google Scholar] [CrossRef]
- Thermo Fisher Scientific. DNA Reference Guide: Sample Preparation and Purification Solutions. 2020. Available online: https://assets.thermofisher.com/TFS-Assets/BID/brochures/dna-isolation-purification-brochure.pdf (accessed on 30 December 2024).
- Zin, O.A.; Neves, L.M.; Cunha, D.P.; Motta, F.L.; Agonigi, B.N.S.; Horovitz, D.D.G.; Almeida, D.C., Jr.; Malacarne, J.; Rodrigues, A.P.S.; Carvalho, A.B.; et al. Genotypic–Phenotypic Correlations of Hereditary Hyperferritinemia-Cataract Syndrome: Case Series of Three Brazilian Families. Int. J. Mol. Sci. 2023, 24, 11876. [Google Scholar] [CrossRef]
- Zin, O.A.; Neves, L.M.; Motta, F.L.; Horovitz, D.D.G.; Guida, L.; Gomes, L.H.F.; Cunha, D.P.; Rodrigues, A.P.S.; Zin, A.A.; Sallum, J.M.F.; et al. Novel Mutation in CRYBB3 Causing Pediatric Cataract and Microphthalmia. Genes 2021, 12, 1069. [Google Scholar] [CrossRef]
- Garcia, M.; Juhos, S.; Larsson, M.; Olason, P.I.; Martin, M.; Eisfeldt, J.; DiLorenzo, S.; Sandgren, J.; De Ståhl, T.D.; Ewels, P.; et al. Sarek: A portable workflow for whole-genome sequencing analysis of germline and somatic variants. F1000Research 2020, 9, 63. [Google Scholar] [CrossRef]
- ABraOM:Brazilian Genomic Variants. Available online: https://abraom.ib.usp.br/ (accessed on 8 January 2024).
- Cat-Map Complete List. Available online: https://cat-map.wustl.edu/ (accessed on 24 September 2024).
- Brooks, S.P.; Ebenezer, N.D.; Poopalasundaram, S.; Lehmann, O.J.; Moore, A.T.; Hardcastle, A.J. Identification of the gene for Nance-Horan syndrome (NHS). J. Med. Genet. 2004, 41, 768–771. [Google Scholar] [CrossRef]
- Burdon, K.P.; McKay, J.D.; Sale, M.M.; Russell-Eggitt, I.M.; Mackey, D.A.; Wirth, M.G.; Elder, J.E.; Nicoll, A.; Clarke, M.P.; FitzGerald, L.M.; et al. Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am. J. Hum. Genet. 2003, 73, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.P.; Coccia, M.; Hao, R.T.; Kanuga, N.; Machesky, L.M.; Bailly, M.; Cheetham, M.E.; Hardcastle, A.J. The Nance–Horan syndrome protein encodes a functional WAVE homology domain (WHD) and is important for co-ordinating actin remodelling and maintaining cell morphology. Hum. Mol. Genet. 2010, 19, 2421–2432. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.V.; Maddala, R. The Role of the Lens Actin Cytoskeleton in Fiber Cell Elongation and Differentiation. Semin. Cell Dev. Biol. 2006, 17, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Sun, H.Y.; Zhu, H.J.; Sun, R.X.; Wang, Y.; Wang, J.N.; Qin, B.; Zhang, W.W.; Ji, J.D. A novel Nance-Horan syndrome mutation identified by next-generation sequencing in a Chinese family. Int. J. Ophthalmol. 2022, 15, 1015–1019. [Google Scholar] [CrossRef]
- Khan, A.O.; Aldahmesh, M.A.; Mohamed, J.Y.; Alkuraya, F.S. Phenotype-genotype Correlation in Potential Female Carriers of X-linked Developmental Cataract (Nance-Horan Syndrome). Ophthalmic Genet. 2012, 33, 89–95. [Google Scholar] [CrossRef]
- Yu, X.; Zhao, Y.; Yang, Z.; Chen, X.; Kang, G. Genetic research on Nance-Horan syndrome caused by a novel mutation in the NHS gene. Gene 2024, 906, 148223. [Google Scholar] [CrossRef]
- Tian, Q.; Li, Y.; Kousar, R.; Guo, H.; Peng, F.; Zheng, F.; Yang, X.; Long, Z.; Tian, R.; Xia, K.; et al. A novel NHS mutation causes Nance-Horan Syndrome in a Chinese family. BMC Med. Genet. 2017, 18, 2. [Google Scholar] [CrossRef]
- Bixler, A.; Higgins, M.; Hartsfiel, J. The NanceHoran syndrome: A rare X- linked ocular-dental trait with expression in heterozygous females. Clin. Genet. 1984, 26, 30–35. [Google Scholar] [CrossRef]
- Vallot, C.; Ouimette, J.-F.; Rougeulle, C. Establishment of X chromosome inactivation and epigenomic features of the inactive X depend on cellular contexts. BioEssays 2016, 38, 869–880. [Google Scholar] [CrossRef]
- Lu, Z.; Carter, A.C.; Chang, H.Y. Mechanistic insights in X-chromosome inactivation. Phil. Trans. R. Soc. B 2017, 372, 20160356. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, E.; Clayton-Smith, J.; Douzgou, S.; Ramsden, S.C.; Ingram, S.; Hall, G.; Hardcastle, C.L.; Fletcher, T.A.; Taylor, R.L.; Ellingford, J.M.; et al. Clinical utility of genetic testing in 201 preschool children with inherited eye disorders. Genet. Med. 2020, 22, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A.; Nussbaum, R.L.; Brewer, E.D. Lowe Syndrome. In GeneReviews ® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Medsinge, A.; Nischall, K. Pediatric cataract: Challenges and future directions. Clin. Ophthalmol. 2015, 9, 77–90. [Google Scholar]
- Kaur, S.; Sukhija, J.; Kumari, K. Commentary: Genetic testing in cases of pediatric cataract. Indian. J. Ophthalmol. 2022, 70, 2623–2624. [Google Scholar] [CrossRef]
- Neves, L.M.; Pinto, M.; Zin, O.A.; Cunha, D.P.; Agonigi, B.N.S.; Motta, F.L.; Gomes, L.H.F.; Horovitz, D.D.G.; Almeida, D.C., Jr.; Malacarne, J.; et al. The cost of genetic diagnosis of suspected hereditary pediatric cataracts with whole-exome sequencing from a middle-income country perspective: A mixed costing analysis. J. Community Genet. 2024, 15, 235–247. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Verma, I.C.; Paliwal, P.; Singh, K. Genetic Testing in Pediatric Ophthalmology. Indian. J. Pediatr. 2017, 85, 228–236. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Male | Female | ||||
|---|---|---|---|---|---|
| Disease Phenotype | II:3 | II:5 | I:2 | II:4 | |
| Ocular features | Pediatric cataract | + | + | + | − |
| Microcornea | + | + | + | − | |
| Corneal opacity | − | + | + | − | |
| Strabismus | + | + | + | − | |
| Nystagmus | + | − | − | − | |
| Facial features | Long−narrow face | − | − | − | − |
| Bulbous nose | − | + | + | − | |
| Anteverted pinnae 1 | + | + | − | − | |
| Dental features | Screwdriver-shaped incisors | + | + | N/A | − |
| Mulberry molars | + | + | N/A | − | |
| Diastema 2 | + | + | N/A | − | |
| Other | Intellectual disability | + | + | − | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zin, O.A.; Neves, L.M.; Motta, F.L.; Junior, D.C.; Cunha, D.P.; Agonigi, B.N.S.; Malacarne, J.; Rodrigues, A.P.S.; Rodrigues, G.D.; Tinoco, M.L.C.; et al. Genotype–Phenotype Correlations of Nance–Horan Syndrome in Male and Female Carriers of a Novel Variant. Genes 2025, 16, 91. https://doi.org/10.3390/genes16010091
Zin OA, Neves LM, Motta FL, Junior DC, Cunha DP, Agonigi BNS, Malacarne J, Rodrigues APS, Rodrigues GD, Tinoco MLC, et al. Genotype–Phenotype Correlations of Nance–Horan Syndrome in Male and Female Carriers of a Novel Variant. Genes. 2025; 16(1):91. https://doi.org/10.3390/genes16010091
Chicago/Turabian StyleZin, Olivia A., Luiza M. Neves, Fabiana L. Motta, Daltro C. Junior, Daniela P. Cunha, Bruna N. S. Agonigi, Jocieli Malacarne, Ana Paula S. Rodrigues, Gabriela D. Rodrigues, Maria Luisa C. Tinoco, and et al. 2025. "Genotype–Phenotype Correlations of Nance–Horan Syndrome in Male and Female Carriers of a Novel Variant" Genes 16, no. 1: 91. https://doi.org/10.3390/genes16010091
APA StyleZin, O. A., Neves, L. M., Motta, F. L., Junior, D. C., Cunha, D. P., Agonigi, B. N. S., Malacarne, J., Rodrigues, A. P. S., Rodrigues, G. D., Tinoco, M. L. C., Horovitz, D. D. G., Carvalho, A. B., Zin, A. A., Vasconcelos, Z. F. M., & Sallum, J. M. F. (2025). Genotype–Phenotype Correlations of Nance–Horan Syndrome in Male and Female Carriers of a Novel Variant. Genes, 16(1), 91. https://doi.org/10.3390/genes16010091