Integration of GWAS and Co-Expression Network Analysis Identified Main Genes Responsible for Nitrogen Uptake Traits in Seedling Waxy Corn


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Phenotype Determination and Analysis
2.3. Genotype Analysis
2.4. Genome-Wide Association Analysis and Candidate Gene Prediction
2.5. Nitrogen-Response Expression Analysis of the Candidate Genes
3. Results
3.1. Identification of Germplasm Resources and Construction of Variation Map of Local Waxy Corn
3.2. Genome-Wide Association Analysis of Nitrogen Uptake Traits
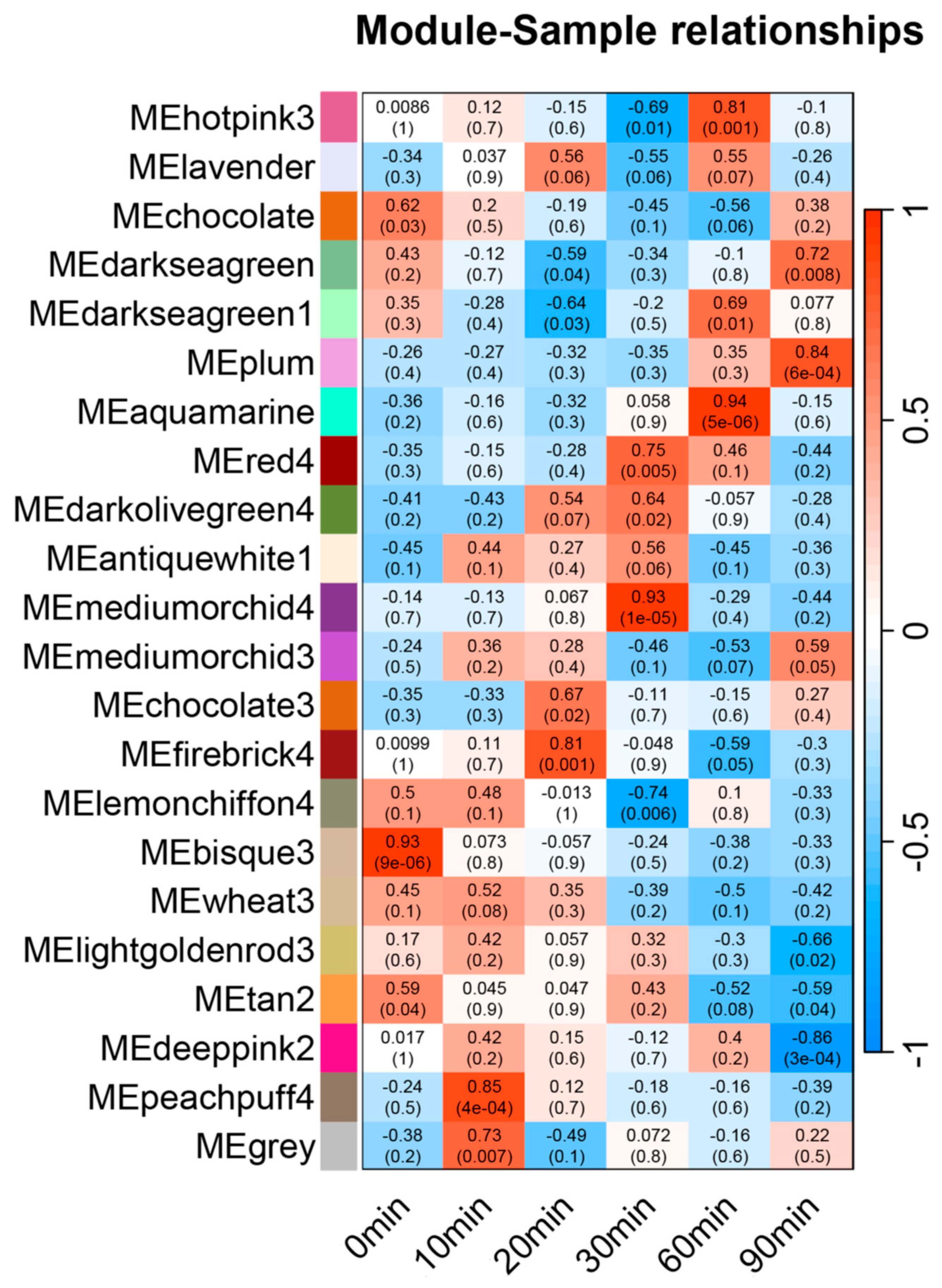
3.3. Transcriptome Co-Expression Network Construction
3.4. Identification of Candidate Genes with Significant Nitrogen Uptake Traits
4. Discussion
4.1. GWAS Was Used to Screen the Candidate Genes Related to Nitrogen Uptake Traits
4.2. Application of WGCNA Co-Expression Network in Candidate Gene Mining
4.3. Analysis of Common Gene Function Discovered by GWAS and WGCNA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dang, D.; Guan, Y.; Zheng, H.; Zhang, X. Genome-wide association study and genomic prediction on plant architecture traits in sweet corn and waxy corn. Plants 2023, 12, 303. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.R.; Lu, B.S.; Shi, Y.X. Dynamics of waxy corn breeding and industry development in China. Maize Sci. 2016, 24, 67–71. (In Chinese) [Google Scholar]
- Li, Y.; Zhong, K.; Wang, X. Sensory evaluation and model prediction of vacuum-packed fresh corn during long-term storage. Foods 2023, 12, 478. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.N.; Reng, B.Z.; Zhao, B. Effects of nitrogen application on filling characteristics and nutritional quality of summer maize. J. Appl. Ecol. 2019, 30, 3771–3776. (In Chinese) [Google Scholar]
- Chen, G.Q.; Zhao, J.Y.; Wang, X. Effects of combined application of base fertilizer N, P and K and nitrogen topdressing at jointing stage on gelatinization characteristics of waxy maize harvested at fresh eating stage. Jiangsu J. Agric. Sci. 2014, 30, 73–79. (In Chinese) [Google Scholar]
- Kurai, T.; Wakayama, M.; Abiko, T. Introduction of the ZmDof1 gene into rice enhances carbon and nitrogen assimilation under low-nitrogen conditions. Plant Biotechnol. J. 2011, 9, 826–837. [Google Scholar] [CrossRef]
- Ge, M.; Wang, Y.; Liu, Y. The NIN-like protein 5 (ZmNLP5) transcription factor is involved in modulating the nitrogen response in maize. Plant J. 2019, 102, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lawit, S.J.; Weers, B. Overexpression of zmm28 increases maize grain yield in the field. Proc. Natl. Acad. Sci. USA 2019, 116, 23850–23858. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, Z.; Li, X. Involvement of a truncated MADS-box transcription factor ZmTMM1 in root nitrate foraging. J. Exp. Bot. 2020, 71, 4547–4561. [Google Scholar] [CrossRef]
- Du, H.; Ning, L.; He, B. Cross-species root transcriptional network analysis highlights conserved modules in response to nitrate between maize and sorghum. Int. J. Mol. Sci. 2020, 21, 1445. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Ye, J.; Yao, X.; Zhao, P.; Xuan, W. Genome-wide associated study identifies NAC42-activated nitrate transporter conferring high nitrogen use efficiency in rice. Nat. Commun. 2019, 10, 5279. [Google Scholar] [CrossRef] [PubMed]
- Júlia, S.M.; Leandro, F.M.; Danilo, H.L.; Giovanni, G.; Miriam, S.V. Association mapping for traits related to nitrogen use efficiency in tropical maize lines under field conditions. Plant Soil 2017, 421, 453–463. [Google Scholar]
- Ma, L.L.; Qing, C.Y.; Frei, U.; Shen, Y.O.; Lubberstedt, T. Association mapping for root system architecture traits under two nitrogen conditions in germplasm enhancement of maize doubled haploid lines. Crop J. 2020, 8, 213–226. [Google Scholar] [CrossRef]
- Ndlovu, N.; Spillane, C.; McKeown, P.C.; Cairns, J.E.; Das, B. Genome-wide association studies of grain yield and quality traits under optimum and low-nitrogen stress in tropical maize (Zea mays L.). Theor. Appl. Genet. 2022, 135, 4351–4370. [Google Scholar] [CrossRef]
- Chen, Z.; Liao, M.; Yang, Z. Co-expression network analysis of genes and networks associated with wheat pistillody. PeerJ 2022, 10, 13902. [Google Scholar] [CrossRef]
- Wang, W. Transcriptome and co-expression network analysis reveals the molecular mechanism of rice root systems in response to low-nitrogen conditions. Int. J. Mol. Sci. 2023, 24, 5290. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Diaz, J.; Beguerisse-Díaz, M.; Poole, P.S. Extracting information from gene coexpression networks of Rhizobium leguminosarum. J. Comput. Biol. 2022, 29, 752–768. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zenda, T.; Dong, A. Global transcriptome and weighted gene co-expression network analyses of growth-stage-specific drought stress responses in maize. Front. Genet. 2021, 12, 645443. [Google Scholar] [CrossRef]
- Luo, B.; Li, J.; Li, B. Mining synergistic genes for nutrient utilization and disease resistance in maize based on co-expression network and consensus QTLs. Front. Plant Sci. 2022, 13, 1013598. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Zhang, J.; Cao, J. Hub gene mining and co-expression network construction of low-temperature response in maize of seedling by WGCNA. Genes 2023, 14, 1598. [Google Scholar] [CrossRef]
- Liu, P.; Jiang, L.; Long, P. A genome-wide co-expression network analysis revealed ZmNRAMP6-mediated regulatory pathway involved in maize tolerance to lead stress. Theor. Appl. Genet. 2023, 136, 122. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; He, C.; Yan, S. A metabolic roadmap of waxy corn flavor. Mol. Plant 2024, 17, 1883–1898. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.K.; Huang, J.L. Experimental principles and techniques of plant physiology and biochemistry. Beijing High. Educ. Press 2015, 55–57. (In Chinese) [Google Scholar]
- Xie, P.J.; Ke, Y.T.; Kuo, L.Y. Modified CTAB protocols for high-molecular-weight DNA extractions from ferns. Appl. Plant Sci. 2023, 3, e11526. [Google Scholar] [CrossRef]
- Slifer, S.H. PLINK: Key functions for data analysis. Curr. Protoc. Hum. Genet. 2018, 97, 59. [Google Scholar] [CrossRef]
- Monier, B.; Casstevens, T.M.; Bradbury, P.J. rTASSEL: An R interface to TASSEL for analyzing genomic diversity. J. Open Source Softw. 2022, 7, 4530. [Google Scholar] [CrossRef]
- Shringarpure, S.S.; Bustamante, C.D.; Lange, K. Efficient analysis of large datasets and sex bias with ADMIXTURE. BMC Bioinform. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Y.W.; Zhang, Z.C.; Xiang, Y.; Liu, M.H.; Zhou, Y.H.; Zuo, J.F.; Zhang, H.Q.; Chen, Y.; Zhang, Y.M.; et al. A compressed variance component mixed model for detecting QTNs, and QTN-by-environment and QTN-by-QTN interactions in genome-wide association studies. Mol. Plant 2022, 4, 630–650. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lin, Y.; Chen, J. Genome-wide association studies provide genetic insights into natural variation of seed-size-related traits in mungbean. Front. Plant Sci. 2022, 13, 997988. [Google Scholar] [CrossRef]
- Chan, B.K.C. Data Analysis Using R Programming. In Biostatistics for Human Genetic Epidemiology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 47–122. [Google Scholar]
- Pablo, C. Variant annotation and functional prediction: SnpEff. Methods Mol. Biol. 2012, 1, 289–314. [Google Scholar]
- Trapnell, C.; Hendrickson, D.G. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B-Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Fernández, A.; Segura-Alabart, N. The MultiFurcating Neighbor-Joining Algorithm for Reconstructing Polytomic Phylogenetic Trees. J. Mol. Evol. 2023, 6, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.W. IIIVmrMLM: The R and C++ tools associated with 3VmrMLM, a comprehensive GWAS method for dissecting quantitative traits. Mol. Plant 2022, 15, 1251–1253. [Google Scholar] [CrossRef]
- Wang, J.E.; Mao, Y.X.; Huang, T.Q. Water and heat stresses during grain formation affect the physicochemical properties of waxy maize starch. J. Sci. Food Agric. 2021, 101, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Karunarathne, S.D.; Han, Y.; Zhang, X.Q. Genome-wide association study and identification of candidate genes for nitrogen use efficiency in barley (Hordeum vulgare L.). Front. Plant Sci. 2020, 11, 571912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhou, T.; Zhang, Y. Genome-wide identification and functional characterization reveals the pivotal roles of BnaA8.ATG8F in salt stress tolerance and nitrogen limitation adaptation in allotetraploid rapeseed. Int. J. Mol. Sci. 2022, 23, 11318. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wang, W.; Chen, J. Genetic improvement toward nitrogen-use efficiency in rice: Lessons and perspectives. Mol. Plant 2023, 16, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Meng, Z.; Yue, R. Genome wide association analysis for yield related traits in maize. BMC Plant Biol. 2022, 22, 449. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Wang, N.; Bao, F. Genome-wide association study reveals genetic basis of trace elements accumulation in maize kernels. Agriculture 2022, 12, 262. [Google Scholar] [CrossRef]
- Li, C.; Huang, Y.; Huang, R. The genetic architecture of amylose biosynthesis in maize kernel. Plant Biotechnol. J. 2018, 16, 688–695. [Google Scholar] [CrossRef]
- Davidson, R.M.; Gowda, M. Comparative transcriptomics of three Poaceae species reveals patterns of gene expression evolution. Plant J. 2012, 71, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Ficklin, S.P.; Feltus, F.A. Gene coexpression network alignment and conservation of gene modules between two grass species: Maize and rice. Plant Physiol. 2011, 156, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, X.; Zhang, D. Genome-wide identification of gene expression in contrasting maize inbred lines under field drought conditions reveals the significance of transcription factors in drought tolerance. PLoS ONE 2017, 12, e0179477. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Wang, F.; Kong, J. Functional analysis of ZmMADS1 a reveals its role in regulating starch biosynthesis in maize endosperm. Sci. Rep. 2019, 9, 3253. [Google Scholar]
- Trung, K.H.; Tran, Q.H.; Bui, N.H. A weak allele of FASCIATED EAR 2 (FEA2) increases maize kernel row number (KRN) and yield in elite maize hybrids. Agronomy 2020, 10, 1774. [Google Scholar] [CrossRef]
- Li, W.; Yu, Y.; Gong, S. Effects of endogenous and exogenous corn protein and its hydrolysates on the structural change and starch digestibility of fried corn starch. Int. J. Food Sci. Technol. 2021, 56, 2732–2741. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, P.; Wang, C. Genome-wide association study uncovers new genetic loci and candidate genes underlying seed chilling-germination in maize. PeerJ 2021, 9, e11707. [Google Scholar] [CrossRef]
- Zhou, X.; Muhammad, I.; Lan, H. Recent advances in the analysis of cold tolerance in maize. Front. Plant Sci. 2022, 13, 866034. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Samayoa, L.F.; Yang, C.J. A conserved genetic architecture among populations of the maize progenitor, teosinte, was radically altered by domestication. Proc. Natl. Acad. Sci. USA 2021, 118, e2112970118. [Google Scholar] [CrossRef]
- Yang, M.; Geng, M.; Shen, P. Effect of post-silking drought stress on the expression profiles of genes involved in carbon and nitrogen metabolism during leaf senescence in maize. Plant Physiol. Biochem. 2019, 135, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Raghuram, N.; Sopory, S.K. Roles of nitrate, nitrite and ammonium ion in phytochrome regulation of nitrate reductase gene expression in maize. IUBMB Life 1999, 47, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Nowak, B.; Tomkowiak, A.; Sobiech, A. Identification and analysis of candidate genes associated with yield structure traits and maize yield using next-generation sequencing technology. Genes 2023, 15, 56. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Guo, Y.; Qiu, L.J. Genome-wide identification and evolutionary analysis of leucine-rich repeat receptor-like protein kinase genes in soybean. BMC Plant Biol. 2016, 16, 58. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Rees, D.C. Glutathione binding to the plant AtAtm3 transporter and implications for the conformational coupling of ABC transporters. eLife 2022, 11, e76140. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Sample Size | Minimum | Maximum | Mean | Median | Standard Deviation | Variance | Skewness | Kurtosis |
|---|---|---|---|---|---|---|---|---|---|
| 2021 | 534 | 437.86 | 3741.43 | 2025.22 | 2130.71 | 785.67 | 617,280.64 | −0.400 | −0.603 |
| 2022 | 534 | 457.46 | 3781 | 2030.19 | 2120.21 | 782.42 | 612,179.10 | −0.354 | −0.572 |
| Blup | 534 | −1402.12 | 1538.34 | 0.0000 | 86.8067 | 695.78 | 484,109.79 | −0.378 | −0.588 |
| Chromosome | Length (bp) | Variance |
|---|---|---|
| chr1 | 301,354,135 | 16,408,388 |
| chr2 | 237,068,873 | 7,902,973 |
| chr3 | 232,140,174 | 12,809,543 |
| chr4 | 241,473,504 | 12,354,858 |
| chr5 | 217,872,852 | 12,993,741 |
| chr6 | 169,174,353 | 11,076,019 |
| chr7 | 176,764,762 | 8,638,318 |
| chr8 | 175,793,759 | 9,368,327 |
| chr9 | 156,750,706 | 9,214,786 |
| chr10 | 150,189,435 | 8,411,799 |
| Total | 2,058,582,553 | 109,178,752 |
| Chromosome | Position (bp) | Trait Name | LOD | r2 (%) | p-Value | GeneID |
|---|---|---|---|---|---|---|
| 1 | 54,929,627 | E1 | 8.13 | 1.94 | 9.46 × 10−10 | Zm00001d029012 |
| multi_env | 8.42 | 0.16 | 4.81 × 10−10 | |||
| 92,818,447 | E1 | 6.40 | 2.62 | 5.64 × 10−8 | ||
| E2 | 8.46 | 3.31 | 4.30 × 10−10 | |||
| multi_env_BLUP | 9.38 | 4.02 | 5.00 × 10−11 | |||
| 198,532,332 | E2 | 8.11 | 2.29 | 9.95 × 10−10 | Zm00001d031678 | |
| multi_env_BLUP | 4.47 | 1.34 | 5.67 × 10−6 | |||
| multi_env | 9.25 | 0.22 | 6.65 × 10−11 | |||
| 231,151,343 | E1 | 5.97 | 1.91 | 1.57 × 10−7 | Zm00001d032578 | |
| E2 | 10.51 | 3.27 | 3.46 × 10−12 | |||
| multi_env_BLUP | 8.46 | 2.83 | 4.36 × 10−10 | |||
| multi_env | 7.79 | 0.20 | 2.09 × 10−9 | |||
| 252,525,197 | E1 | 7.89 | 4.31 | 1.66 × 10−9 | Zm00001d033159 | |
| E2 | 5.75 | 2.92 | 2.65 × 10−7 | |||
| multi_env_BLUP | 10.11 | 5.77 | 8.90 × 10−12 | |||
| 281,763,909 | E1 | 14.25 | 3.60 | 5.42 × 10−16 | Zm00001d034035 | |
| E2 | 13.56 | 3.21 | 2.71 × 10−15 | |||
| multi_env_BLUP | 19.26 | 5.17 | 4.62 × 10−21 | |||
| multi_env | 29.41 | 0.60 | 2.67 × 10−31 | |||
| 286,796,425 | E1 | 7.47 | 3.56 | 4.54 × 10−9 | ||
| E2 | 7.85 | 3.55 | 1.81 × 10−9 | |||
| multi_env_BLUP | 6.08 | 2.96 | 1.22 × 10−7 | |||
| 2 | 241,882,616 | E1 | 11.91 | 1.88 | 1.32 × 10−13 | Zm00001d007890 |
| multi_env_BLUP | 9.80 | 1.57 | 1.85 × 10−11 | |||
| multi_env | 34.07 | 0.45 | 5.40 × 10−36 | |||
| 3 | 105,062,481 | E1 | 9.28 | 1.22 | 6.30 × 10−11 | |
| E2 | 10.29 | 1.29 | 5.84 × 10−12 | |||
| multi_env | 64.55 | 0.81 | 1.31 × 10−66 | |||
| 130,776,903 | E1 | 16.58 | 3.13 | 2.38 × 10−18 | Zm00001d041638 | |
| E2 | 16.10 | 2.86 | 7.32 × 10−18 | |||
| multi_env_BLUP | 18.01 | 3.53 | 8.53 × 10−20 | |||
| multi_env | 97.67 | 1.88 | 8.16 × 10−100 | |||
| 224,502,110 | E1 | 10.01 | 2.28 | 1.12 × 10−11 | Zm00001d044300 | |
| E2 | 13.14 | 2.88 | 7.32 × 10−15 | |||
| multi_env_BLUP | 14.21 | 3.42 | 6.05 × 10−16 | |||
| 5 | 32,772,711 | E1 | 6.04 | 2.01 | 1.33 × 10−7 | Zm00001d014108 |
| multi_env_BLUP | 9.06 | 3.16 | 1.04 × 10−10 | |||
| 75,859,584 | E2 | 5.43 | 1.35 | 5.76 × 10−7 | ||
| multi_env | 89.83 | 2.52 | 5.88 × 10−92 | |||
| 111,565,300 | E1 | 13.54 | 2.76 | 2.86 × 10−15 | ||
| E2 | 11.65 | 2.21 | 2.38 × 10−13 | |||
| multi_env_BLUP | 11.50 | 2.38 | 3.45 × 10−13 | |||
| 6 | 148,266,713 | E1 | 7.48 | 1.92 | 4.33 × 10−9 | Zm00001d038109 |
| E2 | 10.14 | 2.50 | 8.39 × 10−12 | |||
| multi_env | 22.24 | 0.47 | 4.54 × 10−24 | |||
| 166,762,568 | E1 | 12.17 | 3.46 | 7.10 × 10−14 | Zm00001d038905 | |
| E2 | 12.34 | 3.32 | 4.79 × 10−14 | |||
| multi_env_BLUP | 11.90 | 3.48 | 1.33 × 10−13 | |||
| multi_env | 14.49 | 0.32 | 3.14 × 10−16 | |||
| 7 | 3,775,500 | E1 | 7.72 | 3.31 | 2.47 × 10−9 | |
| E2 | 7.95 | 3.23 | 1.43 × 10−9 | |||
| multi_env_BLUP | 7.17 | 3.16 | 9.12 × 10−9 | |||
| 119,463,838 | E1 | 6.09 | 3.33 | 1.20 × 10−7 | Zm00001d020501 | |
| E2 | 8.98 | 4.72 | 1.26 × 10−10 | |||
| multi_env_BLUP | 7.50 | 4.25 | 4.21 × 10−9 | |||
| multi_env | 13.16 | 0.59 | 7.02 × 10−15 | |||
| 144,661,743 | E1 | 6.99 | 2.08 | 1.40 × 10−8 | Zm00001d021167 | |
| E2 | 5.68 | 1.58 | 3.14 × 10−7 | |||
| multi_env | 57.17 | 1.60 | 3.32 × 10−59 | |||
| 165,275,579 | E2 | 9.90 | 1.58 | 1.46 × 10−11 | Zm00001d021877 | |
| multi_env_BLUP | 7.83 | 1.34 | 1.91 × 10−9 | |||
| multi_env | 19.23 | 0.26 | 4.94 × 10−21 | |||
| 177,122,793 | E1 | 9.29 | 5.23 | 6.08 × 10−11 | Zm00001d022414 | |
| E2 | 7.44 | 3.90 | 4.83 × 10−9 | |||
| multi_env_BLUP | 5.84 | 3.30 | 2.17 × 10−7 | |||
| 8 | 70,304,376 | E1 | 26.49 | 5.15 | 2.34 × 10−28 | |
| E2 | 23.36 | 4.19 | 3.32 × 10−25 | |||
| multi_env_BLUP | 22.44 | 4.36 | 2.85 × 10−24 | |||
| multi_env | 15.29 | 0.21 | 4.83 × 10−17 | |||
| 9 | 12,398,673 | E1 | 6.55 | 2.47 | 3.99 × 10−8 | Zm00001d045097 |
| E2 | 6.21 | 2.20 | 8.99 × 10−8 | |||
| multi_env_BLUP | 8.89 | 3.50 | 1.59 × 10−10 | |||
| multi_env | 14.56 | 0.45 | 2.63 × 10−16 | |||
| 10 | 106,059,138 | E1 | 5.58 | 2.98 | 4.02 × 10−7 | Zm00001d025136 |
| multi_env_BLUP | 4.73 | 2.58 | 3.08 × 10−6 |
| ME Names | Main BP | Gene No. | FDR |
|---|---|---|---|
| Aquamarine | Nitrogen compound metabolic process | 1789/5780 | 3.1 × 10−17 |
| Mediumorchild4 | Regulation of nitrogen compound metabolic process | 66/388 | 0.0014 |
| Deeppink2 | Nitrogen compound metabolic process | 764/2556 | 5.6 × 10−5 |
| Peachpuff4 | Cellular macromolecule metabolic process | 13/37 | 0.04 |
| Plum | Single-organism process | 1512/1881 | 2.2 × 10−9 |
| Markers | Gene Name | Chromosome | p-Value | Gene Annotation |
|---|---|---|---|---|
| S1_54929627 | Zm00001d029012 | 1 | 4.81 × 10−10 | Leucine-rich repeat protein kinase family protein |
| S1_198532332 | Zm00001d031678 | 1 | 6.65 × 10−11 | rrb3; retinoblastoma family3 |
| S1_231151343 | Zm00001d032578 | 1 | 2.09 × 10−9 | Dof zinc finger protein DOF1.6 |
| S1_252525197 | Zm00001d033159 | 1 | 8.90 × 10−12 | DEK domain-containing chromatin associated protein |
| S1_281763909 | Zm00001d034035 | 1 | 2.67 × 10−31 | gsht1; glutathione transporter1 |
| S2_241882616 | Zm00001d007890 | 2 | 5.40 × 10−36 | YT521-B-like family protein |
| S3_130776903 | Zm00001d041638 | 3 | 8.16 × 10−100 | |
| S3_224502110 | Zm00001d044300 | 3 | 6.05 × 10−16 | |
| S5_32772711 | Zm00001d014108 | 5 | 5.88 × 10−92 | uce8; ubiquitin conjugating enzyme 8 |
| S6_148266713 | Zm00001d038109 | 6 | 4.54 × 10−24 | |
| S6_166762568 | Zm00001d038905 | 6 | 3.14 × 10−16 | Probable β-14-xylosyltransferase IRX10L |
| S7_119463838 | Zm00001d020501 | 7 | 7.02 × 10−15 | RING/U-box superfamily protein |
| S7_144661743 | Zm00001d021167 | 7 | 3.32 × 10−59 | UDP-glycosyltransferase 74B1 |
| S7_165275579 | Zm00001d021877 | 7 | 4.94 × 10−21 | ak1; adenylyl-sulfate kinase 1 |
| S7_177122793 | Zm00001d022414 | 7 | 2.17 × 10−7 | Ubiquitin carboxyl-terminal hydrolase 24 |
| S9_12398673 | Zm00001d045097 | 9 | 2.63 × 10−16 | Multidrug resistance-associated protein 11 |
| S10_106059138 | Zm00001d025136 | 10 | 3.08 × 10−6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, C.; Dai, H.; Liang, S.; Zhao, H.; Zhou, L. Integration of GWAS and Co-Expression Network Analysis Identified Main Genes Responsible for Nitrogen Uptake Traits in Seedling Waxy Corn. Genes 2025, 16, 126. https://doi.org/10.3390/genes16020126
Luo C, Dai H, Liang S, Zhao H, Zhou L. Integration of GWAS and Co-Expression Network Analysis Identified Main Genes Responsible for Nitrogen Uptake Traits in Seedling Waxy Corn. Genes. 2025; 16(2):126. https://doi.org/10.3390/genes16020126
Chicago/Turabian StyleLuo, Chunmei, Huixue Dai, Shuaiqiang Liang, Han Zhao, and Ling Zhou. 2025. "Integration of GWAS and Co-Expression Network Analysis Identified Main Genes Responsible for Nitrogen Uptake Traits in Seedling Waxy Corn" Genes 16, no. 2: 126. https://doi.org/10.3390/genes16020126
APA StyleLuo, C., Dai, H., Liang, S., Zhao, H., & Zhou, L. (2025). Integration of GWAS and Co-Expression Network Analysis Identified Main Genes Responsible for Nitrogen Uptake Traits in Seedling Waxy Corn. Genes, 16(2), 126. https://doi.org/10.3390/genes16020126